

Regulation of cholesterol and fatty acid synthesis
- PMID: 21504873
- PMCID: PMC3119913
- DOI: 10.1101/cshperspect.a004754
Regulation of cholesterol and fatty acid synthesis
Abstract
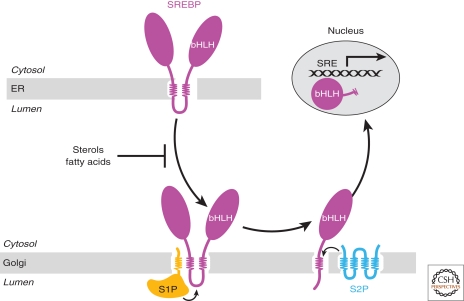
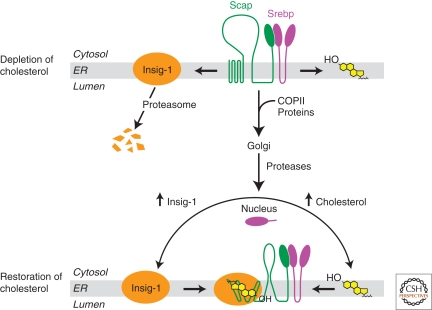
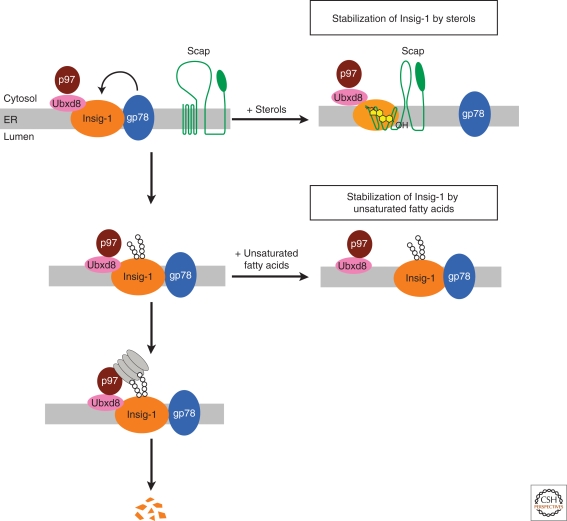
In mammals, intracellular levels of cholesterol and fatty acids are controlled through a feedback regulatory system mediated by a family of transcription factors called sterol regulatory element-binding proteins (SREBPs). SREBPs are synthesized as inactive precursors bound to membranes of the endoplasmic reticulum. When cells are deprived of cholesterol and fatty acids, NH(2)-terminal fragments of SREBPs become proteolytically released from membranes and migrate to the nucleus to activate transcription of genes required for lipid synthesis and uptake. Conversely, lipid repletion inhibits proteolytic processing of SREBPs and thereby suppresses lipid accumulation. We review here studies in cultured cells that reveal the mechanism for regulation of SREBP proteolytic activation, and those in animal models in which SREBP proteolysis has been either activated or inhibited to show the essential role of SREBPs in regulating hepatic lipid homeostasis.
Figures
References
-
- Adams CM, Reitz J, De Brabander JK, Feramisco JD, Li L, Brown MS, Goldstein JL 2004. Cholesterol and 25-Hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and insigs 2. J Biol Chem 279: 52772–52780 - PubMed
-
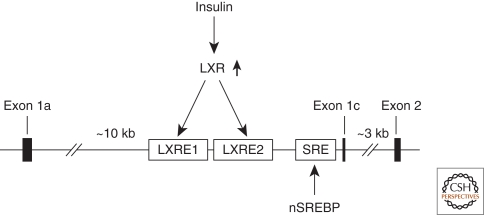
- Amemiya-Kudo M, Shimano H, Yoshikawa T, Yahagi N, Hasty AH, Okazaki H, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, et al. 2000. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol Chem 275: 31078–31085 - PubMed
-
- Brown MS, Goldstein JL 1997. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89: 331–340 - PubMed
-
- Brown MS, Ye J, Rawson RB, Goldstein JL 2000. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell 100: 391–398 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical