

Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain
- PMID: 21505060
- PMCID: PMC3126636
- DOI: 10.1124/jpet.111.180257
Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain
Abstract
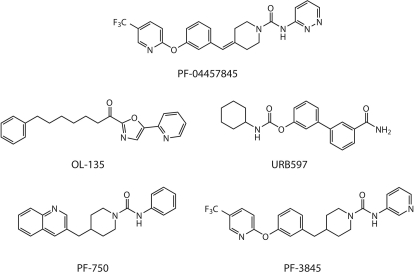
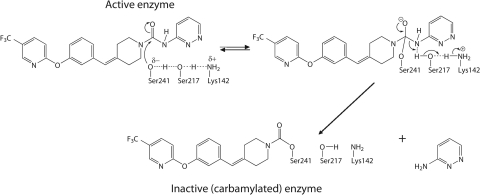
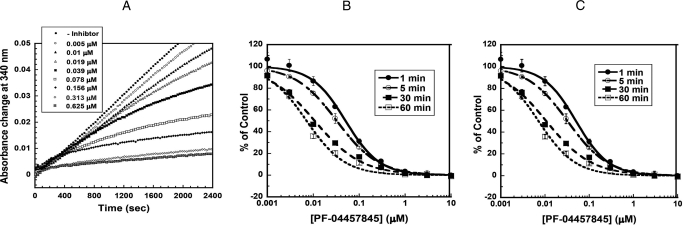
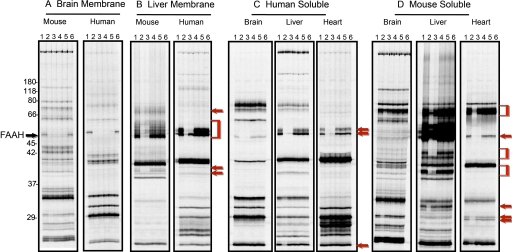
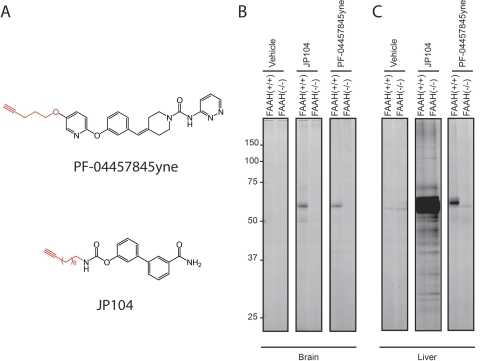
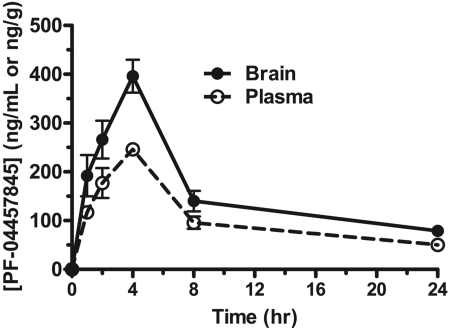
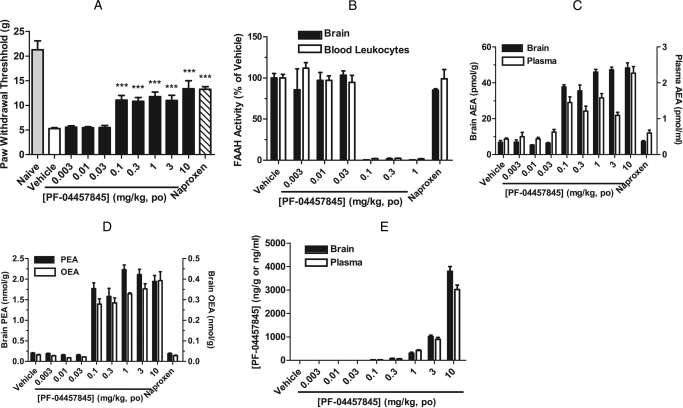
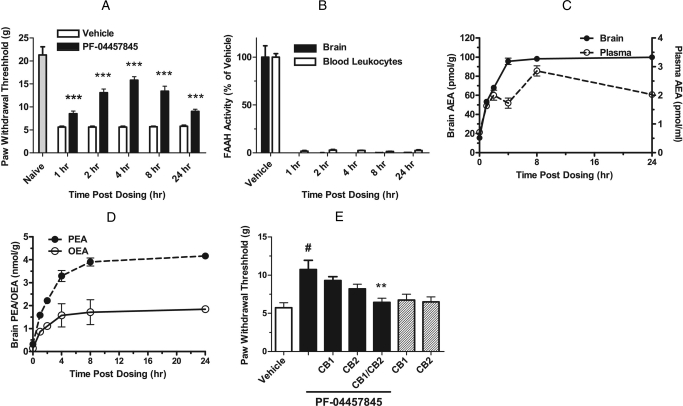
The endogenous cannabinoid (endocannabinoid) anandamide is principally degraded by the integral membrane enzyme fatty acid amide hydrolase (FAAH). Pharmacological blockade of FAAH has emerged as a potentially attractive strategy for augmenting endocannabinoid signaling and retaining the beneficial effects of cannabinoid receptor activation, while avoiding the undesirable side effects, such as weight gain and impairments in cognition and motor control, observed with direct cannabinoid receptor 1 agonists. Here, we report the detailed mechanistic and pharmacological characterization of N-pyridazin-3-yl-4-(3-{[5-(trifluoromethyl)pyridin-2-yl]oxy}benzylidene)piperidine-1-carboxamide (PF-04457845), a highly efficacious and selective FAAH inhibitor. Mechanistic studies confirm that PF-04457845 is a time-dependent, covalent FAAH inhibitor that carbamylates FAAH's catalytic serine nucleophile. PF-04457845 inhibits human FAAH with high potency (k(inact)/K(i) = 40,300 M(-1)s(-1); IC(50) = 7.2 nM) and is exquisitely selective in vivo as determined by activity-based protein profiling. Oral administration of PF-04457845 produced potent antinociceptive effects in both inflammatory [complete Freund's adjuvant (CFA)] and noninflammatory (monosodium iodoacetate) pain models in rats, with a minimum effective dose of 0.1 mg/kg (CFA model). PF-04457845 displayed a long duration of action as a single oral administration at 1 mg/kg showed in vivo efficacy for 24 h with a concomitant near-complete inhibition of FAAH activity and maximal sustained elevation of anandamide in brain. Significantly, PF-04457845-treated mice at 10 mg/kg elicited no effect in motility, catalepsy, and body temperature. Based on its exceptional selectivity and in vivo efficacy, combined with long duration of action and optimal pharmacokinetic properties, PF-04457845 is a clinical candidate for the treatment of pain and other nervous system disorders.
Figures
References
-
- Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, et al. (2007) Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry 46:13019–13030 - PubMed
-
- Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, Hwang I, Hedrick MP, Leung D, Acevedo O, et al. (2005) Discovery of a potent, selective, and efficacious class of reversible α-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J Med Chem 48:1849–1856 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
