

Lafora progressive myoclonus epilepsy: NHLRC1 mutations affect glycogen metabolism
- PMID: 21505799
- PMCID: PMC3154284
- DOI: 10.1007/s00109-011-0758-y
Lafora progressive myoclonus epilepsy: NHLRC1 mutations affect glycogen metabolism
Abstract
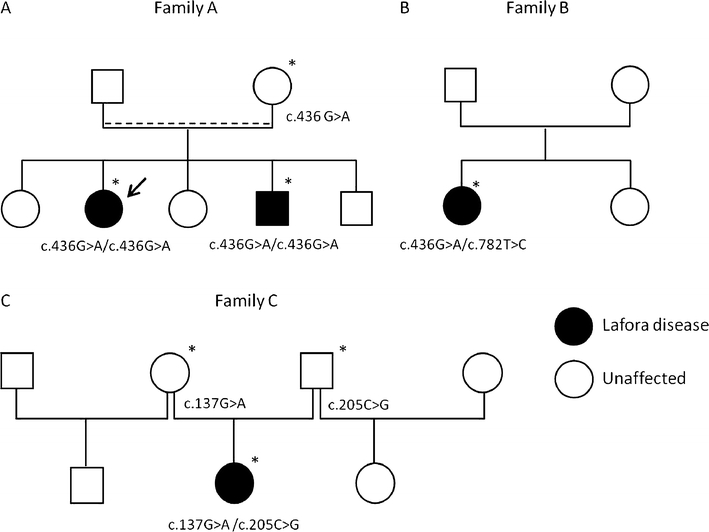
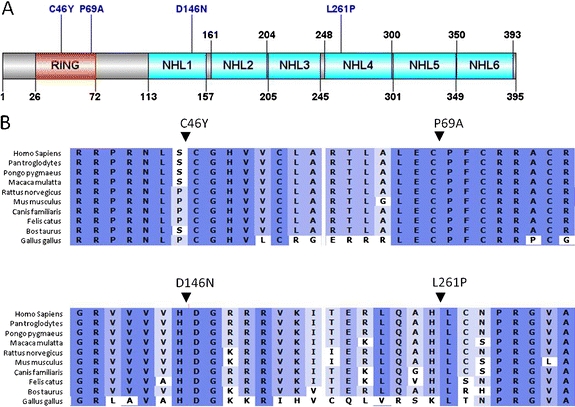
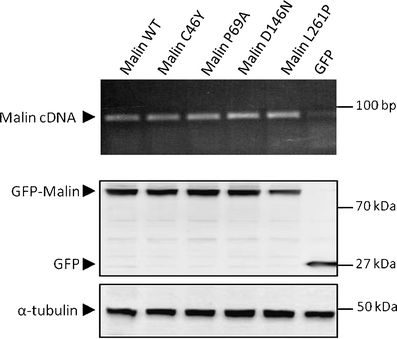
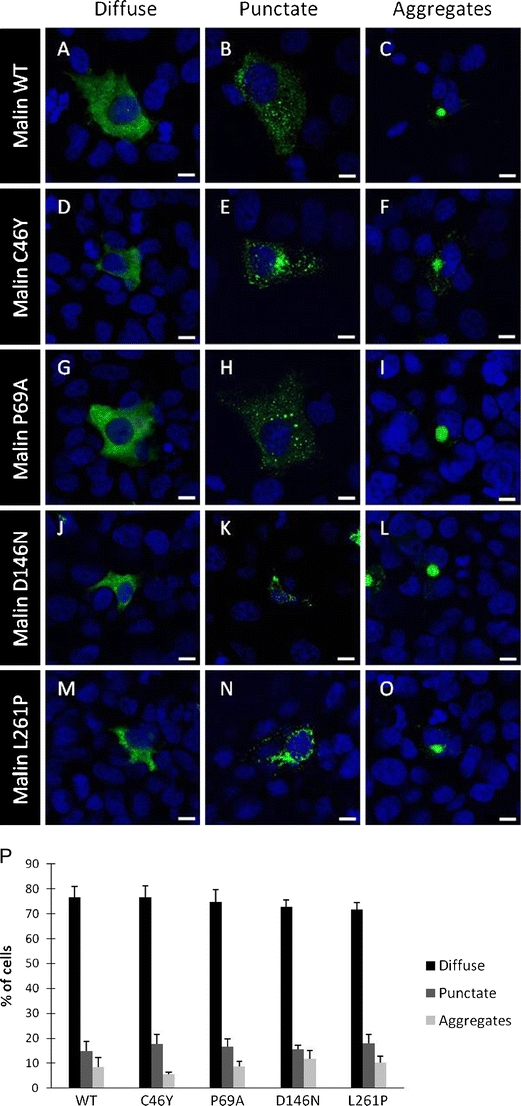
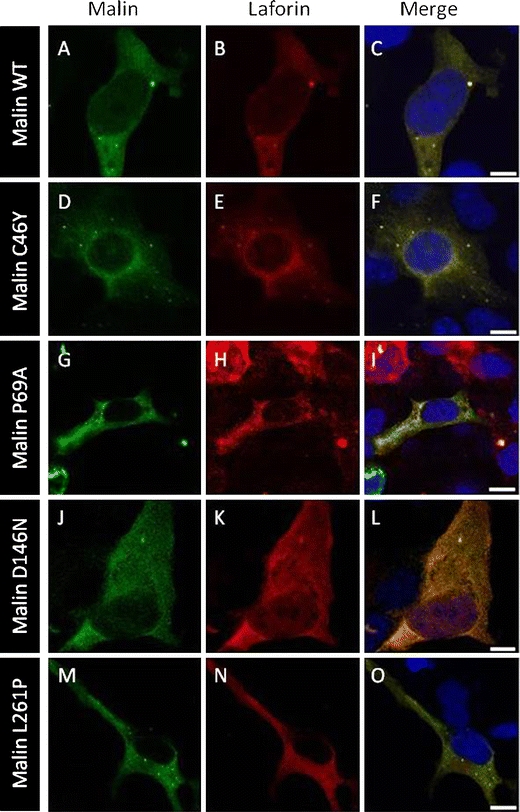
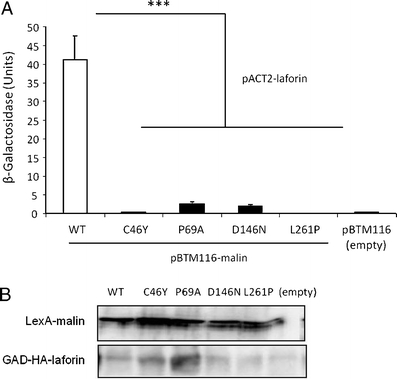
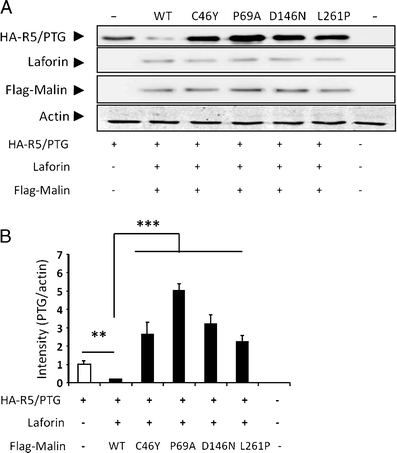
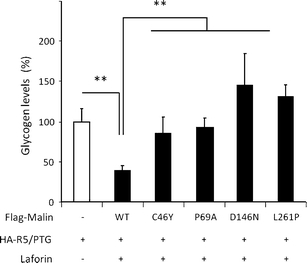
Lafora disease is a fatal autosomal recessive form of progressive myoclonus epilepsy. Patients manifest myoclonus and tonic-clonic seizures, visual hallucinations, intellectual, and progressive neurologic deterioration beginning in adolescence. The two genes known to be involved in Lafora disease are EPM2A and NHLRC1 (EPM2B). The EPM2A gene encodes laforin, a dual-specificity protein phosphatase, and the NHLRC1 gene encodes malin, an E3-ubiquitin ligase. The two proteins interact with each other and, as a complex, are thought to regulate glycogen synthesis. Here, we report three Lafora families with two novel pathogenic mutations (C46Y and L261P) and two recurrent mutations (P69A and D146N) in NHLRC1. Investigation of their functional consequences in cultured mammalian cells revealed that malin(C46Y), malin(P69A), malin(D146N), and malin(L261P) mutants failed to downregulate the level of R5/PTG, a regulatory subunit of protein phosphatase 1 involved in glycogen synthesis. Abnormal accumulation of intracellular glycogen was observed with all malin mutants, reminiscent of the polyglucosan inclusions (Lafora bodies) present in patients with Lafora disease.
Figures
References
-
- Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, Lindhout D, Augustijn PB, Tassinari CA, Malafosse RM, Topcu M, et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2) Hum Mol Genet. 1999;8:345–352. doi: 10.1093/hmg/8.2.345. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
