

Ciliopathies
- PMID: 21506742
- PMCID: PMC3640822
- DOI: 10.1056/NEJMra1010172
Ciliopathies
Abstract
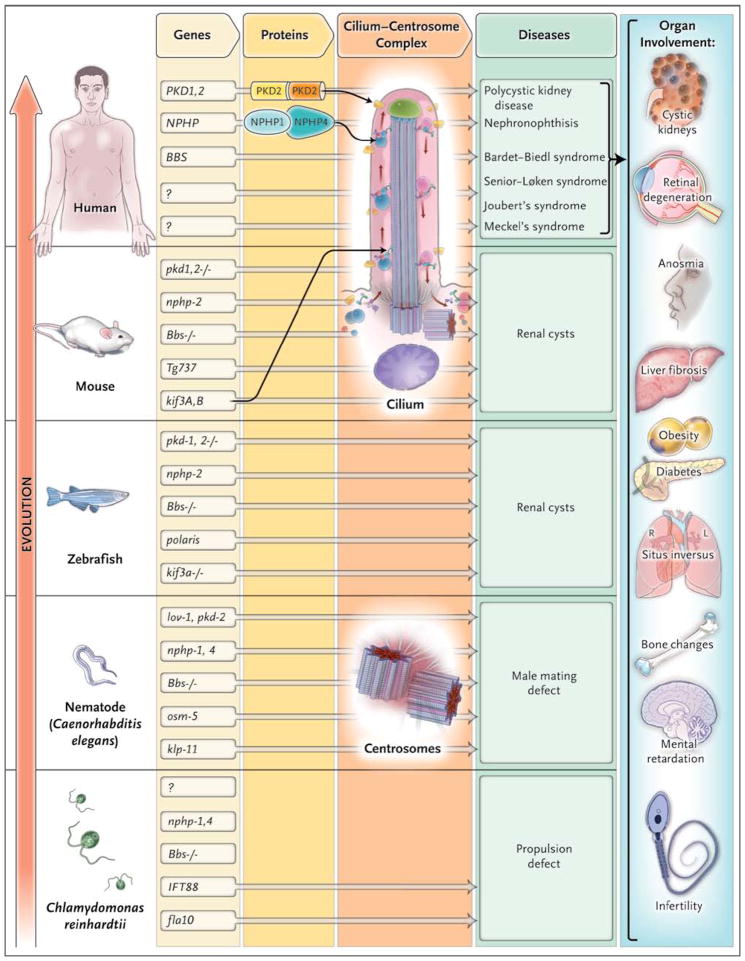
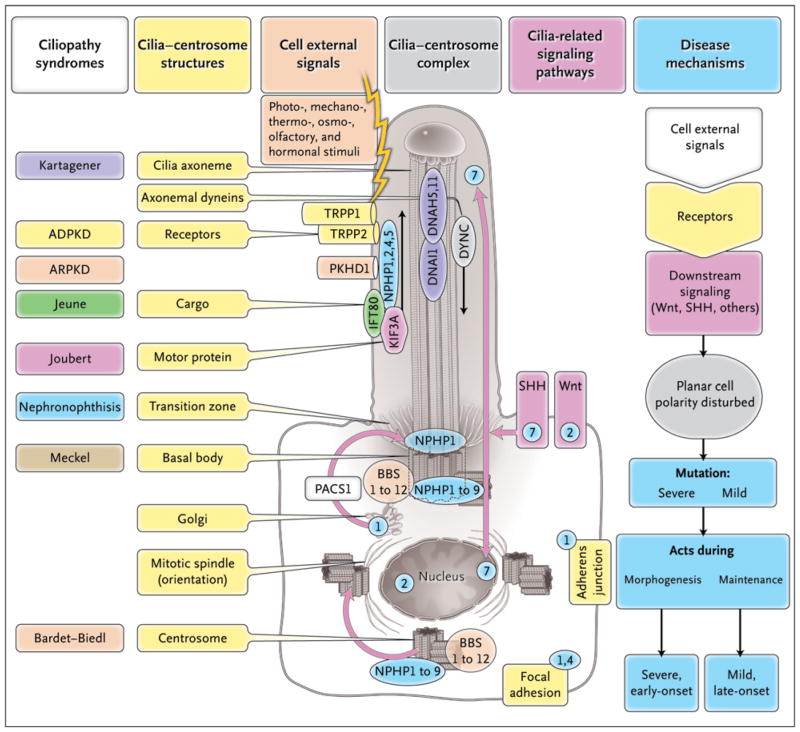
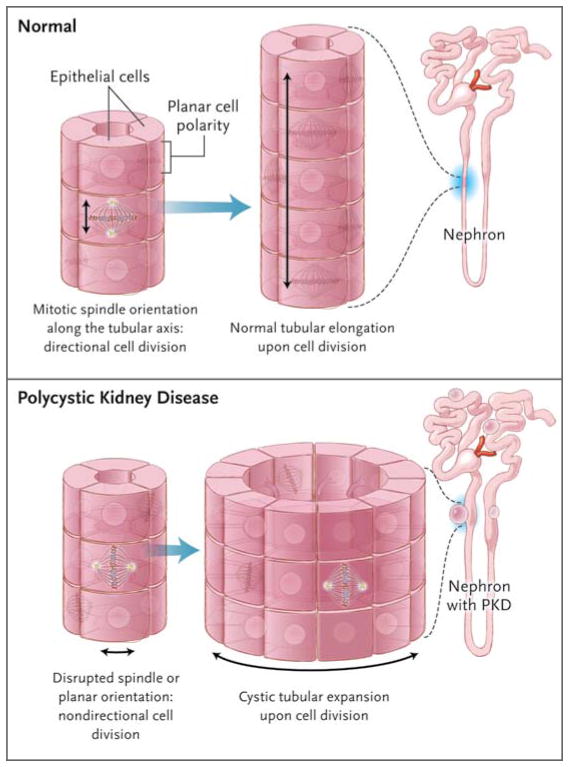
Diverse developmental and degenerative single-gene disorders such as polycystic kidney disease, nephronophthisis, retinitis pigmentosa, the Bardet–Biedl syndrome, the Joubert syndrome, and the Meckel syndrome may be categorized as ciliopathies — a recent concept that describes diseases characterized by dysfunction of a hairlike cellular organelle called the cilium. Most of the proteins that are altered in these single-gene disorders function at the level of the cilium–centrosome complex, which represents nature’s universal system for cellular detection and management of external signals. Cilia are microtubule-based structures found on almost all vertebrate cells. They originate from a basal body, a modified centrosome, which is the organelle that forms the spindle poles during mitosis. The important role that the cilium–centrosome complex plays in the normal function of most tissues appears to account for the involvement of multiple organ systems in ciliopathies. In this review, we consider the role of the cilium in disease.
Conflict of interest statement
Dr. Benzing reports receiving lecture fees from Novartis, Amgen, and Shire; and Dr. Katsanis, lecture fees from the Beijing Genomics Institute. No other potential conflict of interest relevant to this article was reported.
Figures
References
-
- Fliegauf M, Horvath J, von Schnakenburg C, et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol. 2006;17:2424–33. - PubMed
-
- Bisgrove BW, Yost HJ. The roles of cilia in developmental disorders and disease. Development. 2006;133:4131–43. - PubMed
-
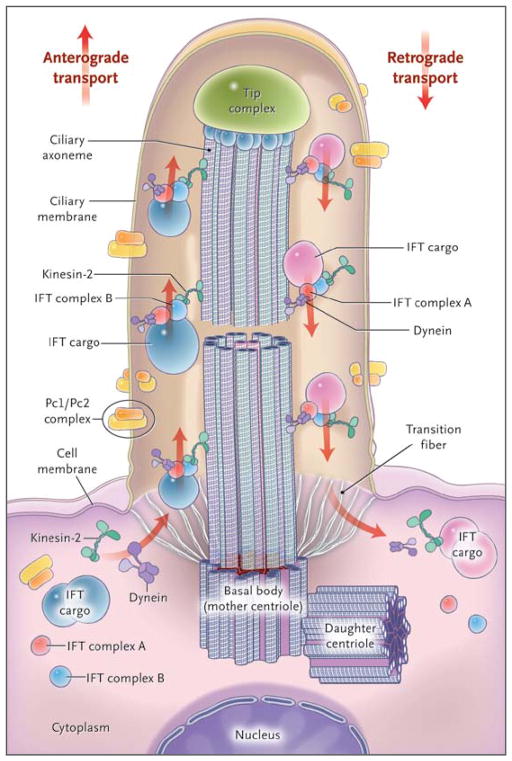
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources