

Observations in APP bitransgenic mice suggest that diffuse and compact plaques form via independent processes in Alzheimer's disease
- PMID: 21514441
- PMCID: PMC3081149
- DOI: 10.1016/j.ajpath.2011.01.052
Observations in APP bitransgenic mice suggest that diffuse and compact plaques form via independent processes in Alzheimer's disease
Abstract
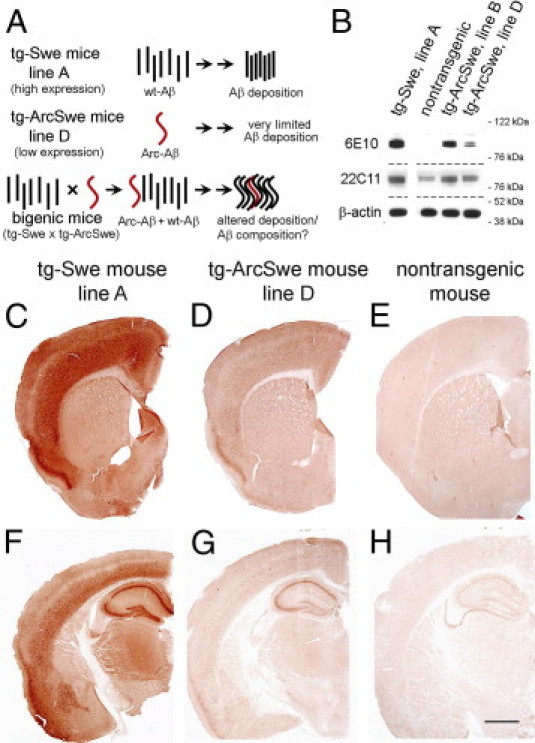
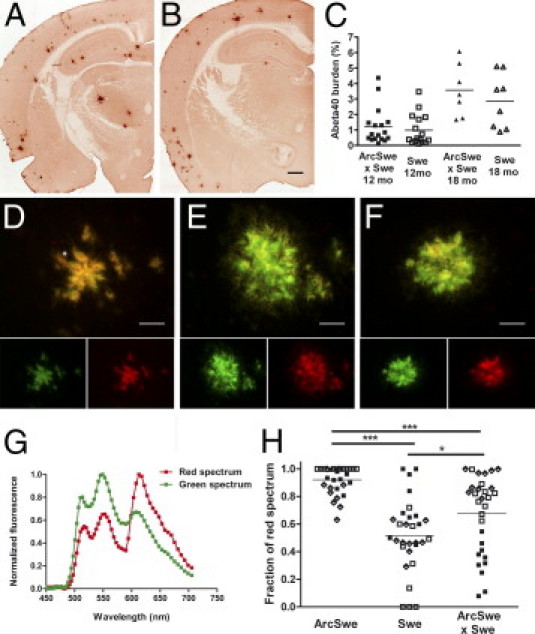
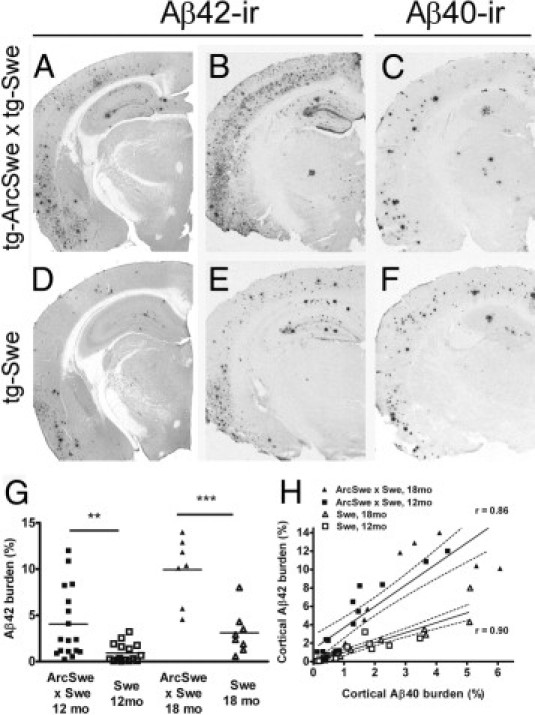
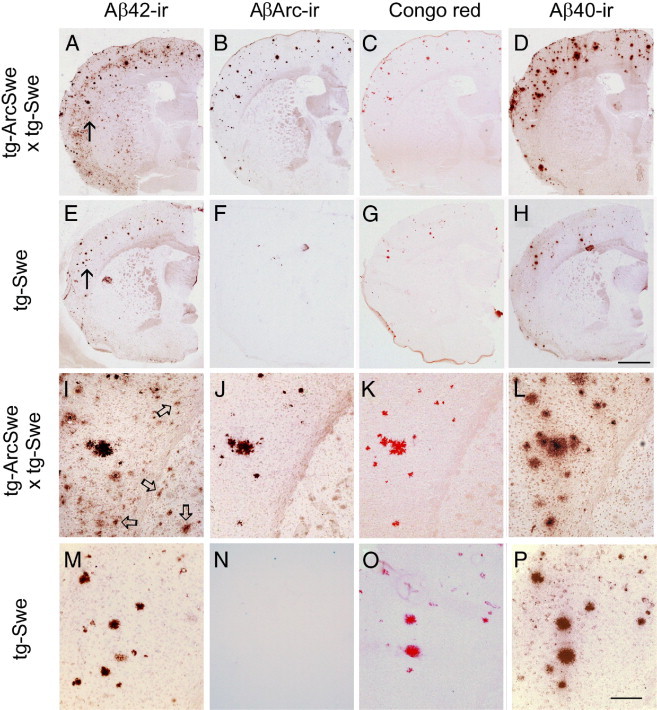
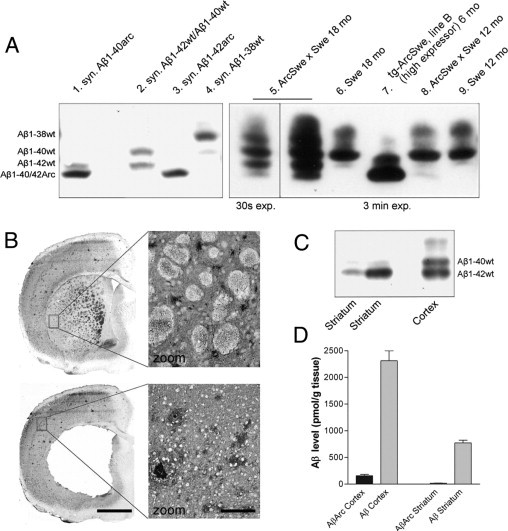
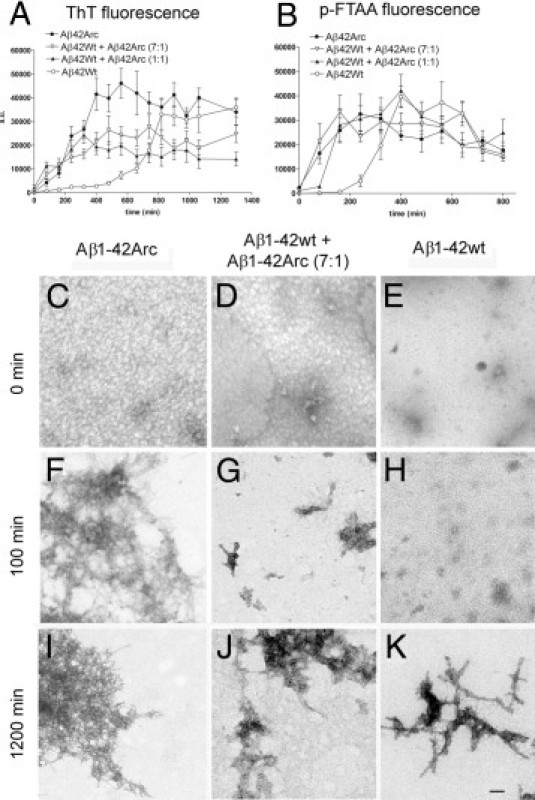
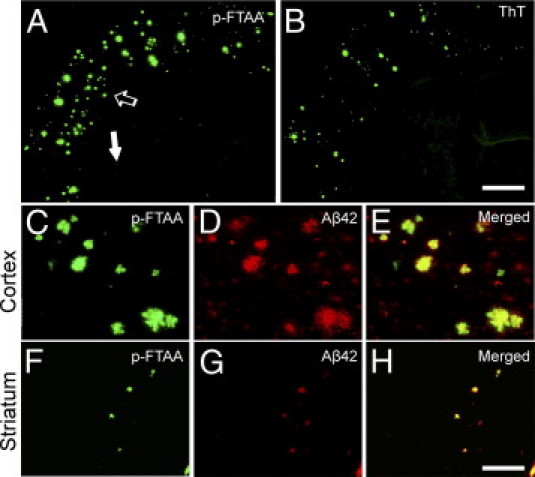
Studies of familial Alzheimer's disease suggest that misfolding and aggregation of amyloid-β (Aβ) peptides initiate the pathogenesis. The Arctic mutation of Aβ precursor protein (APP) results in AD, and Arctic Aβ is more prone to form Aβ protofibrils and extracellular deposits. Herein is demonstrated that the burden of diffuse Aβ deposits but not compact plaques is increased when tg-Swe mice are crossed with tg-ArcSwe mice synthesizing low levels of Arctic Aβ. The diffuse deposits in bitransgenic mice, which contain primarily wild-type Aβ42, accumulate in regions both with and without transgene expression. However, APP processing, when compared with tg-Swe, remains unchanged in young bitransgenic mice, whereas wild-type Aβ42 aggregation is accelerated and fibril architecture is altered in vitro and in vivo when a low level of Arctic Aβ42 is introduced. Thus, the increased number of diffuse deposits is likely due to physical interactions between Arctic Aβ and wild-type Aβ42. The selective increase of a single type of parenchymal Aβ deposit suggests that different pathways lead to formation of diffuse and compact plaques. These findings could have general implications for Alzheimer's disease pathogenesis and particular relevance to patients heterozygous for the Arctic APP mutation. Moreover, it further illustrates how Aβ neuropathologic features can be manipulated in vivo by mechanisms similar to those originally conceptualized in prion research.
Copyright © 2011 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. - PubMed
-
- Nilsberth C., Westlind-Danielsson A., Eckman C.B., Condron M.M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D.B., Younkin S.G., Näslund J., Lannfelt L. The “Arctic” APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–893. - PubMed
-
- Johansson A.S., Berglind-Dehlin F., Karlsson G., Edwards K., Gellerfors P., Lannfelt L. Physiochemical characterization of the Alzheimer's disease–related peptides A beta 1-42Arctic and A beta 1-42wt. FEBS J. 2006;273:2618–2630. - PubMed
-
- Lord A., Kalimo H., Eckman C., Zhang X.Q., Lannfelt L., Nilsson L.N. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
