

Pompe disease gene therapy
- PMID: 21518733
- PMCID: PMC3095055
- DOI: 10.1093/hmg/ddr174
Pompe disease gene therapy
Abstract
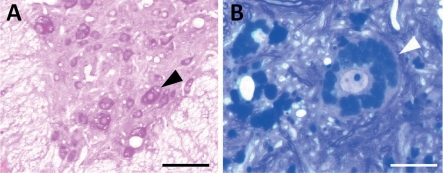
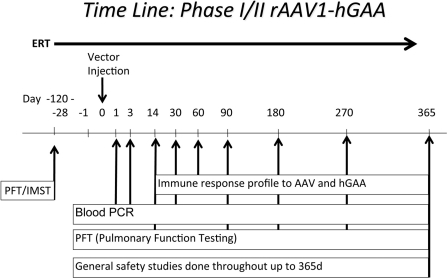
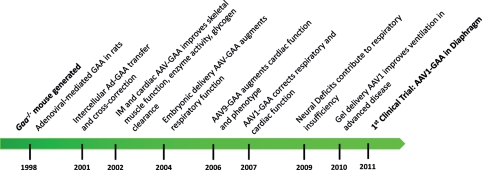
Pompe disease is an autosomal recessive metabolic myopathy caused by the deficiency of the lysosomal enzyme acid alpha-glucosidase and results in cellular lysosomal and cytoplasmic glycogen accumulation. A wide spectrum of disease exists from hypotonia and severe cardiac hypertrophy in the first few months of life due to severe mutations to a milder form with the onset of symptoms in adulthood. In either condition, the involvement of several systems leads to progressive weakness and disability. In early-onset severe cases, the natural history is characteristically cardiorespiratory failure and death in the first year of life. Since the advent of enzyme replacement therapy (ERT), the clinical outcomes have improved. However, it has become apparent that a new natural history is being defined in which some patients have substantial improvement following ERT, while others develop chronic disability reminiscent of the late-onset disease. In order to improve on the current clinical outcomes in Pompe patients with diminished clinical response to ERT, we sought to address the cause and potential for the treatment of disease manifestations which are not amenable to ERT. In this review, we will focus on the preclinical studies that are relevant to the development of a gene therapy strategy for Pompe disease, and have led to the first clinical trial of recombinant adeno-associated virus-mediated gene-based therapy for Pompe disease. We will cover the preliminary laboratory studies and rationale for a clinical trial, which is based on the treatment of the high rate of respiratory failure in the early-onset patients receiving ERT.
Figures
References
-
- Kishnani P.S., Hwu W.L., Mandel H., Nicolino M., Yong F., Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006;148:671–676. - PubMed
-
- Van der Beek N.A., Hagemans M.L., Reuser A.J., Hop W.C., Van der Ploeg A.T., Van Doorn P.A., Wokke J.H. Rate of disease progression during long-term follow-up of patients with late-onset Pompe disease. Neuromusc. Disord. 2009;19:113–117. - PubMed
-
- Byrne B.J., Kishnani P.S., Case L.E., Muller-Felber W., Prasad S., van der Ploeg A.T. Pompe Disease: Design, methodology, and early findings from the Pompe Registry. Mol. Genet. Metab. 2011 in press. - PubMed
-
- Kishnani P.S., Howell R.R. Pompe disease in infants and children. J. Pediatr. 2004;144(Suppl. 5):S35–43. - PubMed
-
- Raben N., Nichols R.C., Boerkoel C., Plotz P. Genetic defects in patients with glycogenosis type II (acid maltase deficiency) Muscle Nerve. 1995;3:S70–74. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
