

Mutations in lipoprotein lipase that block binding to the endothelial cell transporter GPIHBP1
- PMID: 21518912
- PMCID: PMC3093490
- DOI: 10.1073/pnas.1100992108
Mutations in lipoprotein lipase that block binding to the endothelial cell transporter GPIHBP1
Abstract
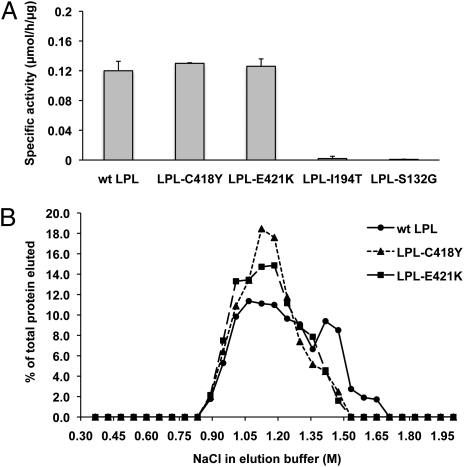
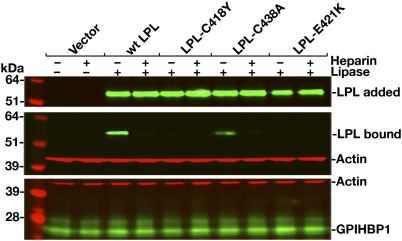
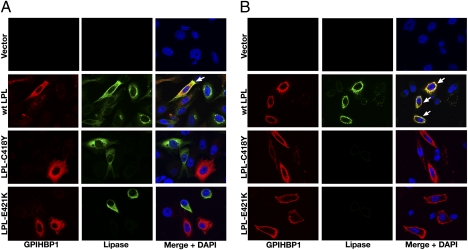
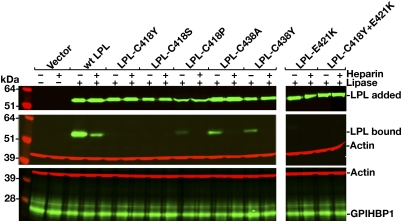
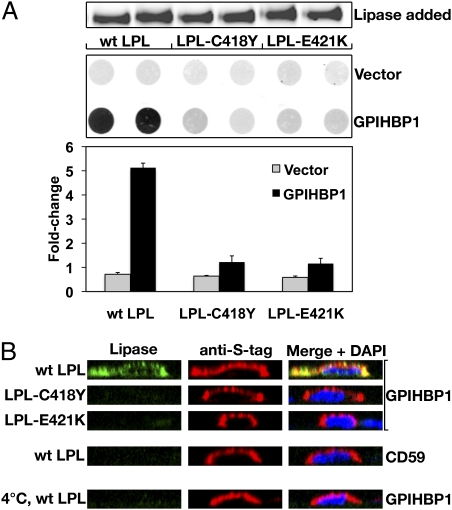
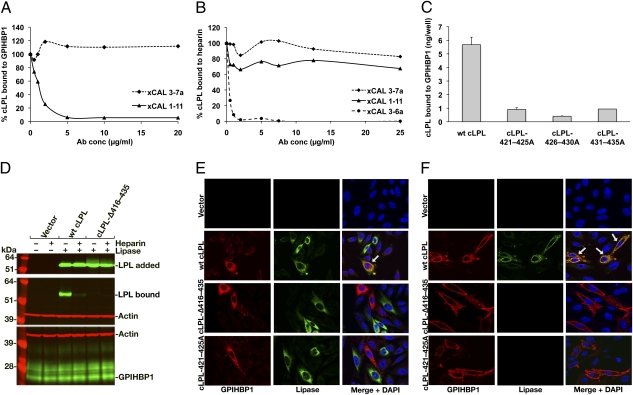
GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells, shuttles lipoprotein lipase (LPL) from subendothelial spaces to the capillary lumen. An absence of GPIHBP1 prevents the entry of LPL into capillaries, blocking LPL-mediated triglyceride hydrolysis and leading to markedly elevated triglyceride levels in the plasma (i.e., chylomicronemia). Earlier studies have established that chylomicronemia can be caused by LPL mutations that interfere with catalytic activity. We hypothesized that some cases of chylomicronemia might be caused by LPL mutations that interfere with LPL's ability to bind to GPIHBP1. Any such mutation would provide insights into LPL sequences required for GPIHBP1 binding. Here, we report that two LPL missense mutations initially identified in patients with chylomicronemia, C418Y and E421K, abolish LPL's ability to bind to GPIHBP1 without interfering with LPL catalytic activity or binding to heparin. Both mutations abolish LPL transport across endothelial cells by GPIHBP1. These findings suggest that sequences downstream from LPL's principal heparin-binding domain (amino acids 403-407) are important for GPIHBP1 binding. In support of this idea, a chicken LPL (cLPL)-specific monoclonal antibody, xCAL 1-11 (epitope, cLPL amino acids 416-435), blocks cLPL binding to GPIHBP1 but not to heparin. Also, changing cLPL residues 421 to 425, 426 to 430, and 431 to 435 to alanine blocks cLPL binding to GPIHBP1 without inhibiting catalytic activity. Together, these data define a mechanism by which LPL mutations could elicit disease and provide insights into LPL sequences required for binding to GPIHBP1.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Chylomicronemia mutations yield new insights into interactions between lipoprotein lipase and GPIHBP1.Hum Mol Genet. 2012 Jul 1;21(13):2961-72. doi: 10.1093/hmg/dds127. Epub 2012 Apr 6. Hum Mol Genet. 2012. PMID: 22493000 Free PMC article.
-
An LPL-specific monoclonal antibody, 88B8, that abolishes the binding of LPL to GPIHBP1.J Lipid Res. 2016 Oct;57(10):1889-1898. doi: 10.1194/jlr.M070813. Epub 2016 Aug 5. J Lipid Res. 2016. PMID: 27494936 Free PMC article.
-
GPIHBP1 missense mutations often cause multimerization of GPIHBP1 and thereby prevent lipoprotein lipase binding.Circ Res. 2015 Feb 13;116(4):624-32. doi: 10.1161/CIRCRESAHA.116.305085. Epub 2014 Nov 11. Circ Res. 2015. PMID: 25387803 Free PMC article.
-
GPIHBP1, an endothelial cell transporter for lipoprotein lipase.J Lipid Res. 2011 Nov;52(11):1869-84. doi: 10.1194/jlr.R018689. Epub 2011 Aug 15. J Lipid Res. 2011. PMID: 21844202 Free PMC article. Review.
-
Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 and the intravascular processing of triglyceride-rich lipoproteins.J Intern Med. 2012 Dec;272(6):528-40. doi: 10.1111/joim.12003. Epub 2012 Nov 1. J Intern Med. 2012. PMID: 23020258 Free PMC article. Review.
Cited by
-
Lipoprotein Lipase (LPL) Polymorphism and the Risk of Coronary Artery Disease: A Meta-Analysis.Int J Environ Res Public Health. 2017 Jan 16;14(1):84. doi: 10.3390/ijerph14010084. Int J Environ Res Public Health. 2017. PMID: 28275220 Free PMC article. Review.
-
New wrinkles in lipoprotein lipase biology.Curr Opin Lipidol. 2012 Feb;23(1):35-42. doi: 10.1097/MOL.0b013e32834d0b33. Curr Opin Lipidol. 2012. PMID: 22123668 Free PMC article. Review.
-
We FRET so You Don't Have To: New Models of the Lipoprotein Lipase Dimer.Biochemistry. 2018 Jan 16;57(2):241-254. doi: 10.1021/acs.biochem.7b01009. Epub 2018 Jan 5. Biochemistry. 2018. PMID: 29303250 Free PMC article.
-
Multimerization of glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) and familial chylomicronemia from a serine-to-cysteine substitution in GPIHBP1 Ly6 domain.J Biol Chem. 2014 Jul 11;289(28):19491-9. doi: 10.1074/jbc.M114.558528. Epub 2014 May 20. J Biol Chem. 2014. PMID: 24847059 Free PMC article. Clinical Trial.
-
Biochemistry and pathophysiology of intravascular and intracellular lipolysis.Genes Dev. 2013 Mar 1;27(5):459-84. doi: 10.1101/gad.209296.112. Genes Dev. 2013. PMID: 23475957 Free PMC article. Review.
References
-
- Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency. In: Scriver CR, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th Ed. Vol 2. New York: McGraw-Hill; 2001. pp. 2789–2816.
-
- Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: Genetics, lipid uptake, and regulation. J Lipid Res. 2002;43:1997–2006. - PubMed
-
- Wang H, Eckel RH. Lipoprotein lipase: From gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–E288. - PubMed
-
- Hayden MR, Ma Y, Brunzell J, Henderson HE. Genetic variants affecting human lipoprotein and hepatic lipases. Curr Opin Lipidol. 1991;2:104–109.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
