

G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia
- PMID: 21518983
- PMCID: PMC3113597
- DOI: 10.1161/CIRCULATIONAHA.110.988642
G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia
Abstract
Background: Alterations in cardiac energy metabolism downstream of neurohormonal stimulation play a crucial role in the pathogenesis of heart failure. The chronic adrenergic stimulation that accompanies heart failure is a signaling abnormality that leads to the upregulation of G protein-coupled receptor kinase 2 (GRK2), which is pathological in the myocyte during disease progression in part owing to uncoupling of the β-adrenergic receptor system. In this study, we explored the possibility that enhanced GRK2 expression and activity, as seen during heart failure, can negatively affect cardiac metabolism as part of its pathogenic profile.
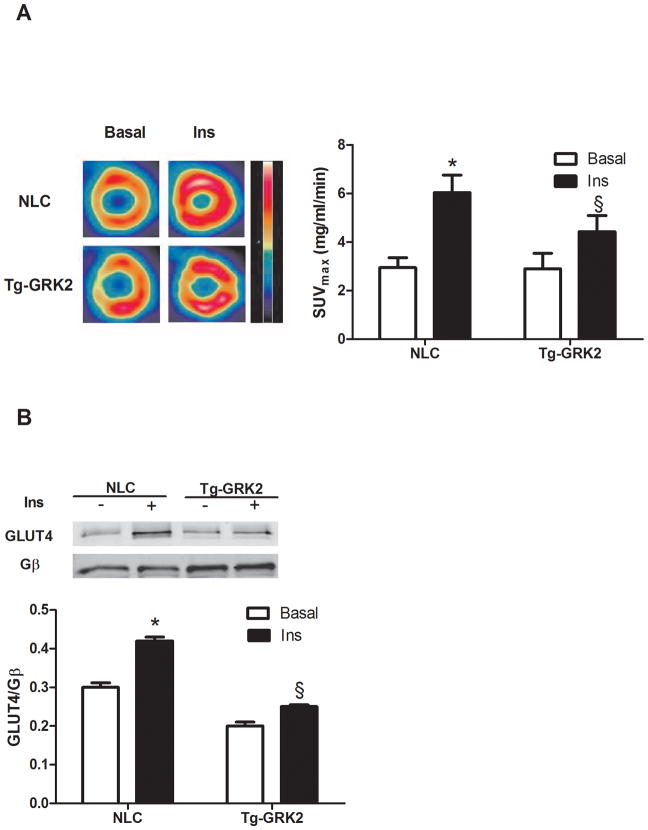
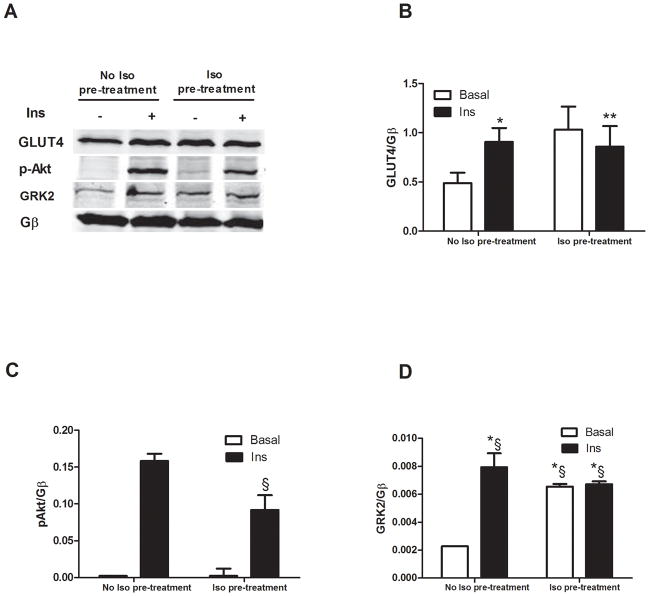
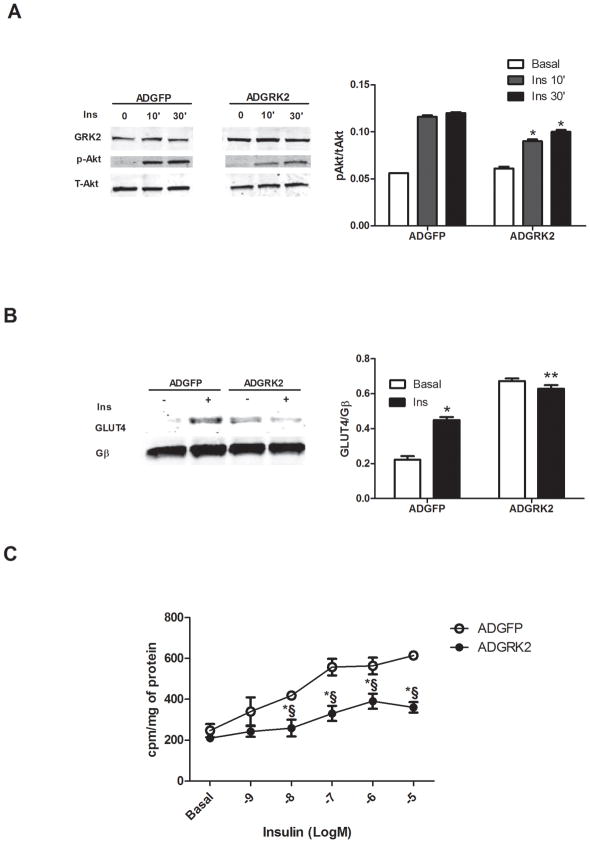
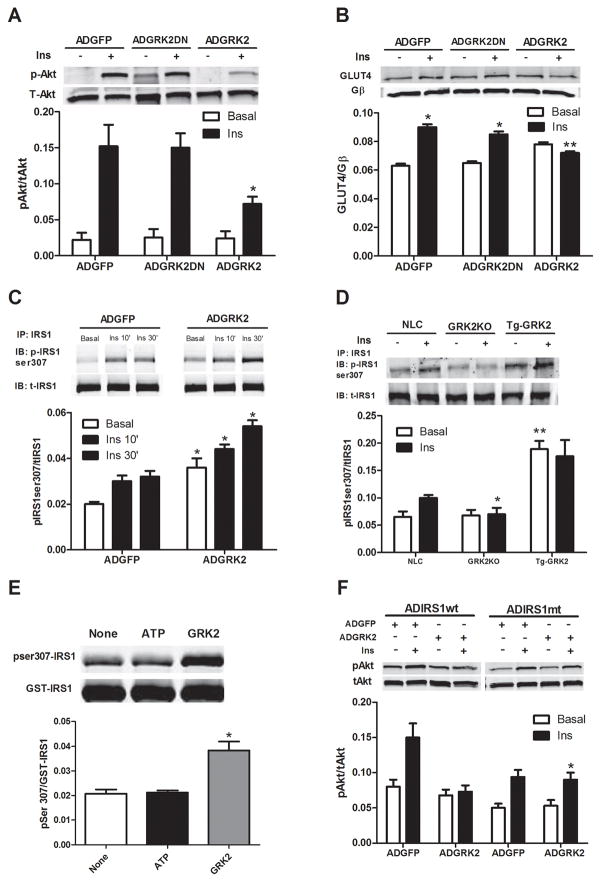
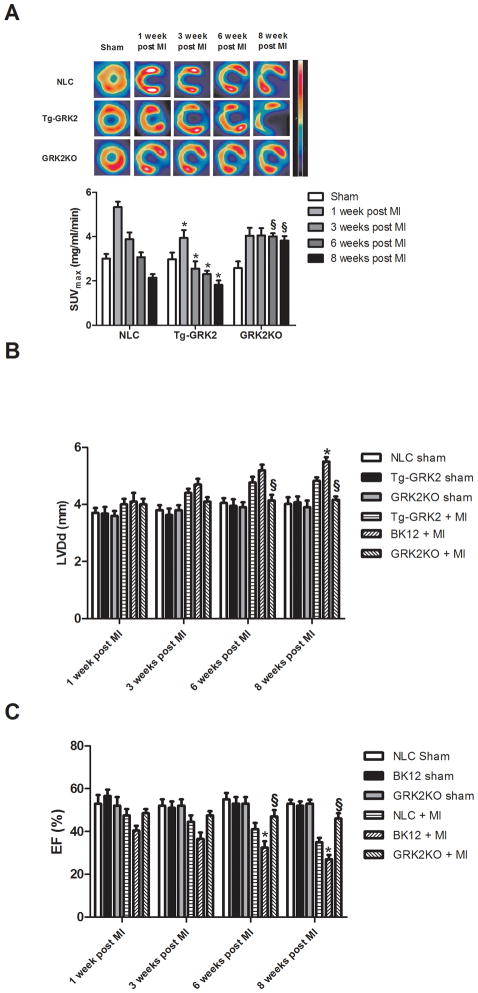
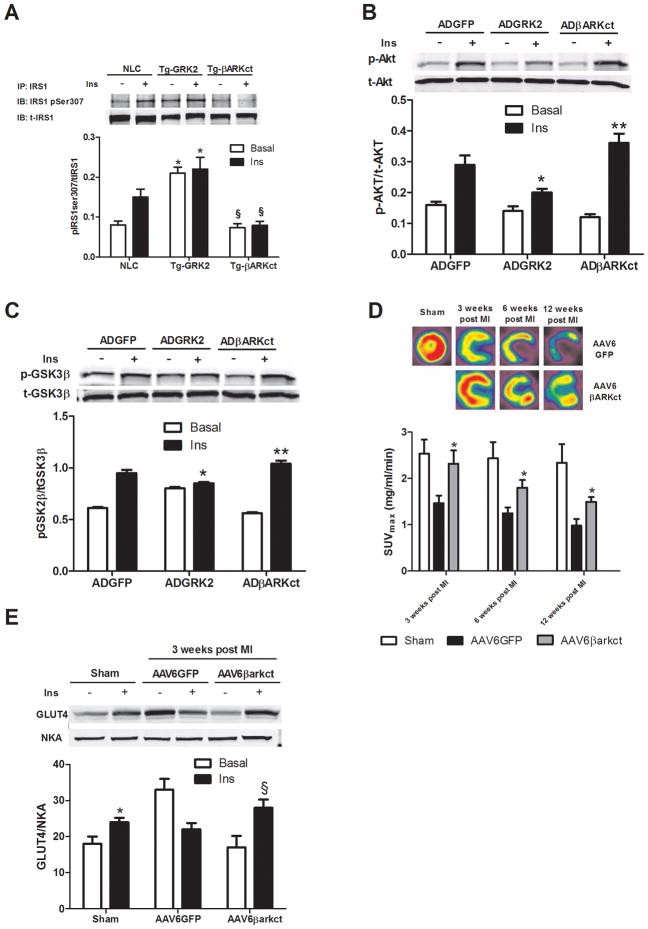
Methods and results: Positron emission tomography studies revealed in transgenic mice that cardiac-specific overexpression of GRK2 negatively affected cardiac metabolism by inhibiting glucose uptake and desensitization of insulin signaling, which increases after ischemic injury and precedes heart failure development. Mechanistically, GRK2 interacts with and directly phosphorylates insulin receptor substrate-1 in cardiomyocytes, causing insulin-dependent negative signaling feedback, including inhibition of membrane translocation of the glucose transporter GLUT4. This identifies insulin receptor substrate-1 as a novel nonreceptor target for GRK2 and represents a new pathological mechanism for this kinase in the failing heart. Importantly, inhibition of GRK2 activity prevents postischemic defects in myocardial insulin signaling and improves cardiac metabolism via normalized glucose uptake, which appears to participate in GRK2-targeted prevention of heart failure.
Conclusions: Our data provide novel insights into how GRK2 is pathological in the injured heart. Moreover, it appears to be a critical mechanistic link within neurohormonal crosstalk governing cardiac contractile signaling/function through β-adrenergic receptors and metabolism through the insulin receptor.
Figures
References
-
- Braunwald E. The Denolin lecture. Congestive heart failure: a half century perspective. Eur Heart J. 2001;22:825–836. - PubMed
-
- Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–2018. - PubMed
-
- Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. - PubMed
-
- Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Cir Res. 2004;95:135–145. - PubMed
-
- Ashrafian H, Frenneaux MP. Metabolic modulation in heart failure: the coming of age. Cardiovasc Drugs Ther. 2007;21:5–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
