

Neuronal cell death in neonatal hypoxia-ischemia
- PMID: 21520238
- PMCID: PMC4000313
- DOI: 10.1002/ana.22419
Neuronal cell death in neonatal hypoxia-ischemia
Abstract
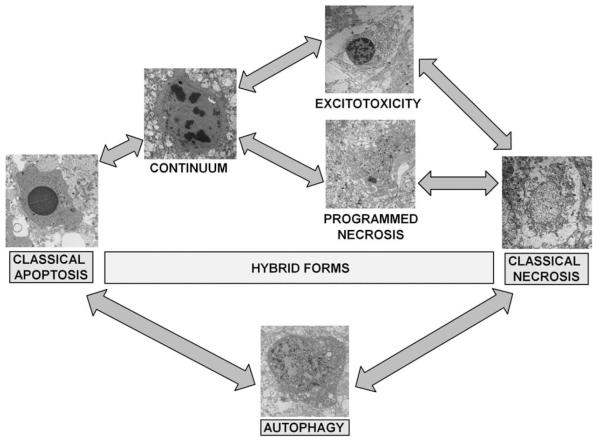
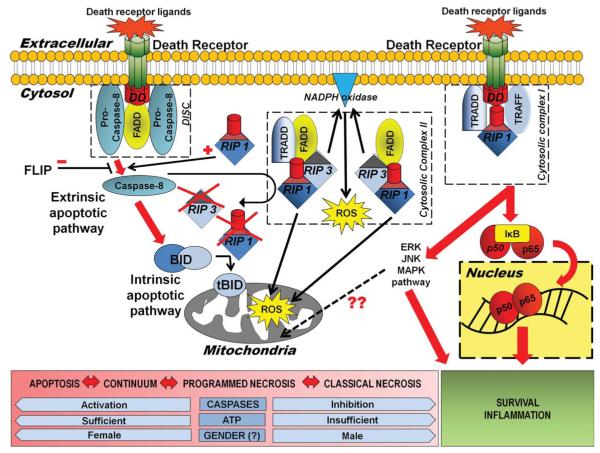
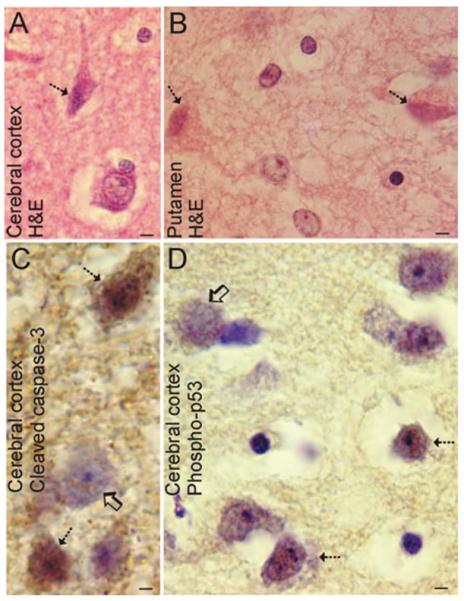
Perinatal hypoxic-ischemic encephalopathy (HIE) is a significant cause of mortality and morbidity in infants and young children. Therapeutic opportunities are very limited for neonatal and pediatric HIE. Specific neural systems and populations of cells are selectively vulnerable in HIE; however, the mechanisms of degeneration are unresolved. These mechanisms involve oxidative stress, excitotoxicity, inflammation, and the activation of several different cell death pathways. Decades ago the structural and mechanistic basis of the cellular degeneration in HIE was thought to be necrosis. Subsequently, largely due to advances in cell biology and to experimental animal studies, emphasis has been switched to apoptosis or autophagy mediated by programmed cell death (PCD) mechanisms as important forms of degeneration in HIE. We have conceptualized based on morphological and biochemical data that this degeneration is better classified according to an apoptosis-necrosis cell death continuum and that programmed cell necrosis has prominent contribution in the neurodegeneration of HIE in animal models. It is likely that neonatal HIE evolves through many cell death chreodes influenced by the dynamic injury landscape. The relevant injury mechanisms remain to be determined in human neonatal HIE, though preliminary work suggests a complexity in the cell death mechanisms greater than that anticipated from experimental animal models. The accurate identification of the various cell death chreodes and their mechanisms unfolding within the immature brain matrix could provide fresh insight for developing meaningful therapies for neonatal and pediatric HIE.
Copyright © 2011 American Neurological Association.
Figures
References
-
- Folkerth RD. Neuropathologic substrate of cerebral palsy. J Child Neurol. 2005;20:940–949. - PubMed
-
- Towfighi J, Zec N, Yager J, et al. Temporal evolution of neuropathologic changes in an immature rat model of cerebral hypoxia: a light microscopic study. Acta Neuropathol. 1995;90:375–386. - PubMed
-
- Myers RE. Four patterns of perinatal brain damage and their conditions of occurrence in primates. Adv Neurol. 1975;10:223–234. - PubMed
-
- Adamsons K, Myers RE. Perinatal asphyxia, causes, detection and neurologic sequelae. Pediatr Clin North Am. 1973;20:465–480. - PubMed
-
- Carloni S, Carnevali A, Cimino M, Balduini W. Extended role of necrotic cell death after hypoxia-ischemia-induced neurodegeneration in the neonatal rat. Neurobiol Dis. 2007;27:354–361. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 NS060703/NS/NINDS NIH HHS/United States
- R01 NS034100/NS/NINDS NIH HHS/United States
- R01 NS079348/NS/NINDS NIH HHS/United States
- R21 NS059529/NS/NINDS NIH HHS/United States
- NS 059529/NS/NINDS NIH HHS/United States
- R01 NS045059/NS/NINDS NIH HHS/United States
- R01 NS065895/NS/NINDS NIH HHS/United States
- R01 NS052098/NS/NINDS NIH HHS/United States
- AG 016282/AG/NIA NIH HHS/United States
- R01 AG016282/AG/NIA NIH HHS/United States
- NS 060703/NS/NINDS NIH HHS/United States
- R01 HD070996/HD/NICHD NIH HHS/United States
- NS 052098/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
