A large-scale benchmark study of existing algorithms for taxonomy-independent microbial community analysis
- PMID: 21525143
- PMCID: PMC3251834
- DOI: 10.1093/bib/bbr009
A large-scale benchmark study of existing algorithms for taxonomy-independent microbial community analysis
Abstract
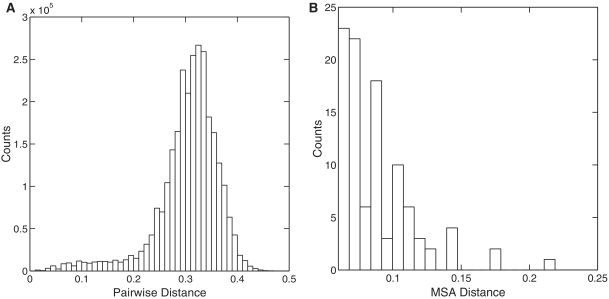
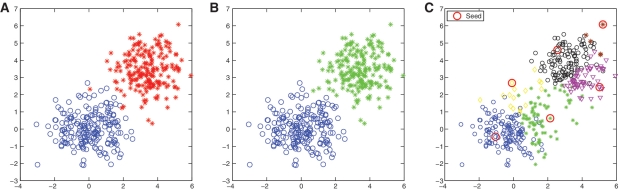
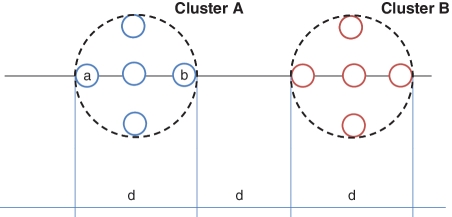
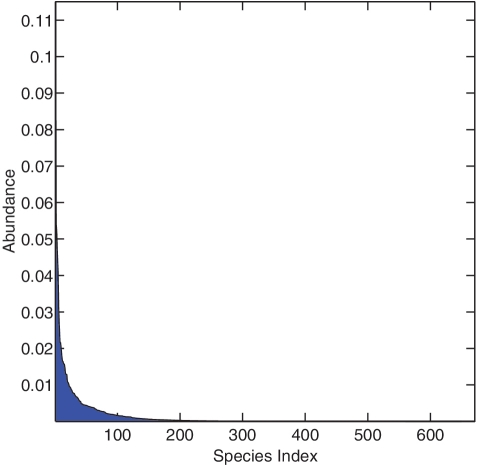
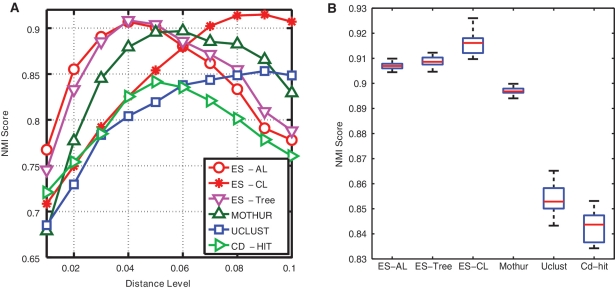
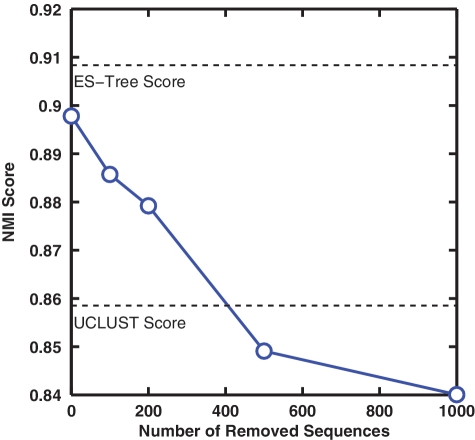
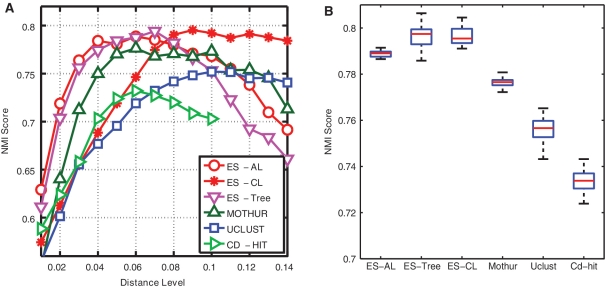
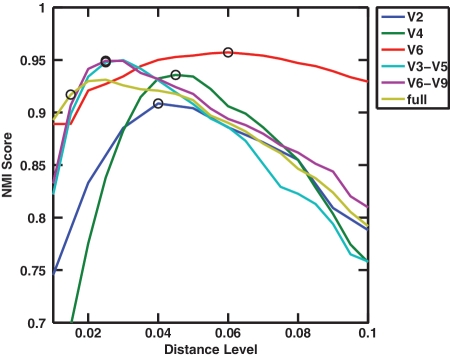
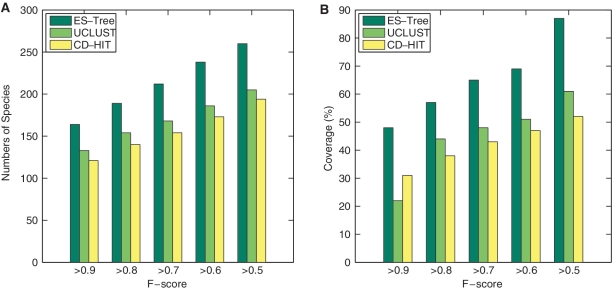
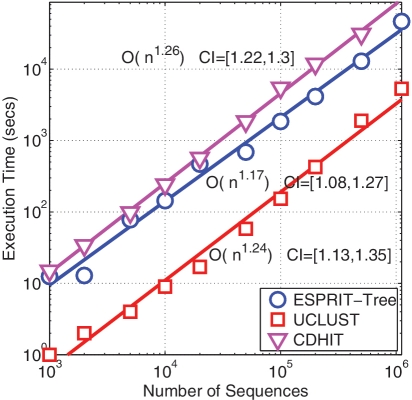
Recent advances in massively parallel sequencing technology have created new opportunities to probe the hidden world of microbes. Taxonomy-independent clustering of the 16S rRNA gene is usually the first step in analyzing microbial communities. Dozens of algorithms have been developed in the last decade, but a comprehensive benchmark study is lacking. Here, we survey algorithms currently used by microbiologists, and compare seven representative methods in a large-scale benchmark study that addresses several issues of concern. A new experimental protocol was developed that allows different algorithms to be compared using the same platform, and several criteria were introduced to facilitate a quantitative evaluation of the clustering performance of each algorithm. We found that existing methods vary widely in their outputs, and that inappropriate use of distance levels for taxonomic assignments likely resulted in substantial overestimates of biodiversity in many studies. The benchmark study identified our recently developed ESPRIT-Tree, a fast implementation of the average linkage-based hierarchical clustering algorithm, as one of the best algorithms available in terms of computational efficiency and clustering accuracy.
Figures