

Delayed cytosolic exposure of Japanese encephalitis virus double-stranded RNA impedes interferon activation and enhances viral dissemination in porcine cells
- PMID: 21525349
- PMCID: PMC3126492
- DOI: 10.1128/JVI.00233-11
Delayed cytosolic exposure of Japanese encephalitis virus double-stranded RNA impedes interferon activation and enhances viral dissemination in porcine cells
Abstract
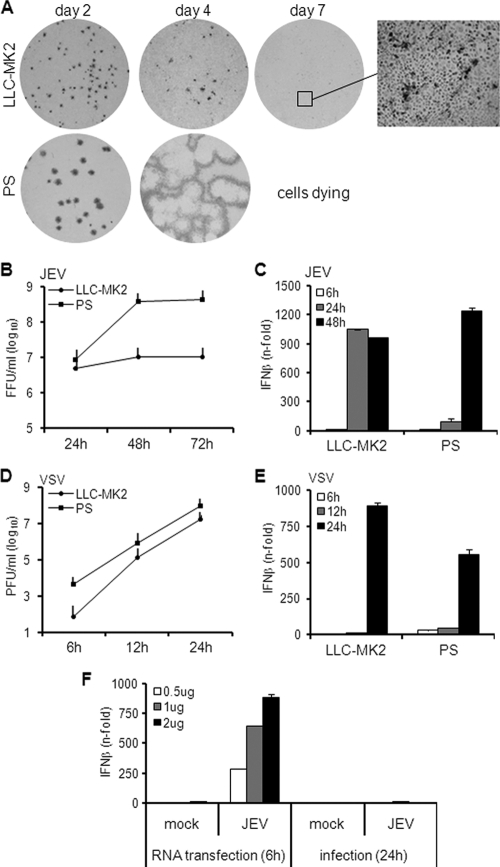
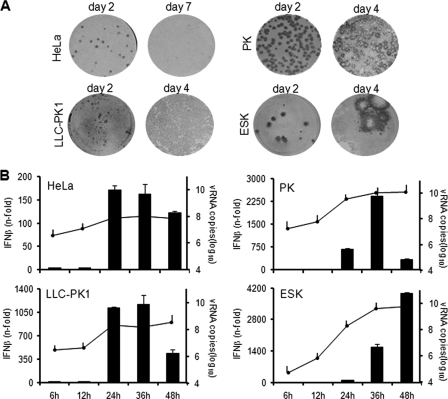
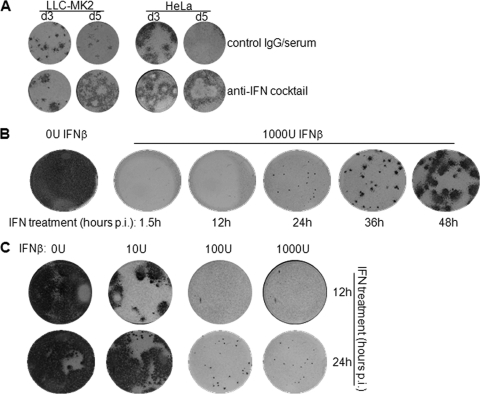
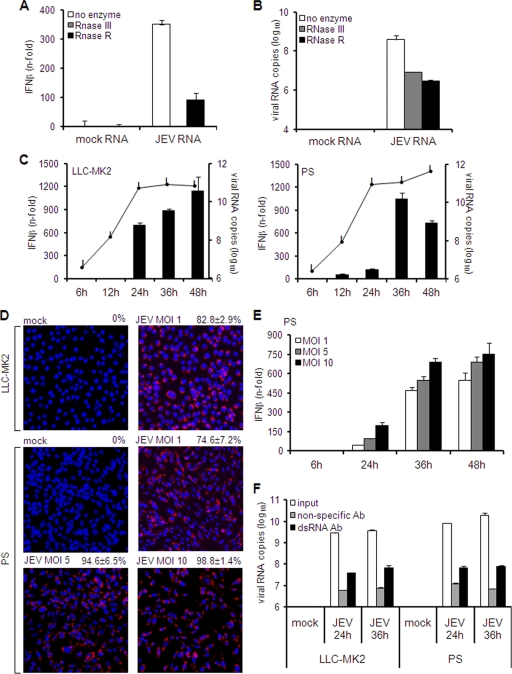
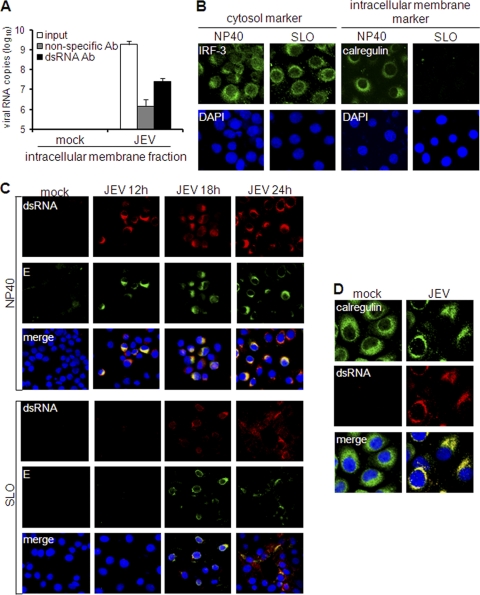
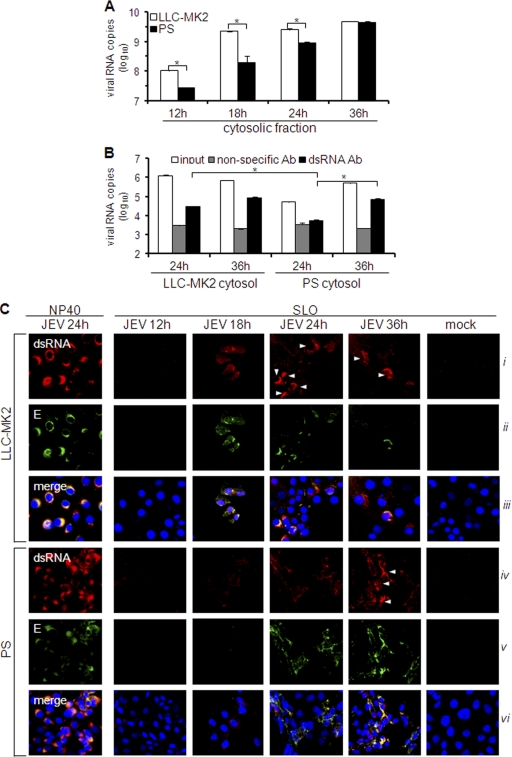
Interferon is a principal component of the host antiviral defense system. In this study, abortive focus formation by Japanese encephalitis virus (JEV) in primate cells was accompanied by early interferon induction, while productive focus formation in porcine cells was associated with a late interferon response. Neutralization antibodies against interferon relieved the restricted infection in primate cells, and increasingly larger foci were generated as treatment with exogenous interferon was delayed, thereby establishing a solid correlation between interferon response and viral dissemination. However, delayed interferon induction in JEV-infected porcine cells occurred in the absence of active inhibition by the virus. We further demonstrated that JEV mediates interferon activation through double-stranded RNA and cytosolic pattern recognition receptors. Immunofluorescence and subcellular fractionation studies revealed that double-stranded RNA is concealed in intracellular membranes at an early phase of infection but eventually appears in the cytosol at later periods, which could then allow detection by cytosolic pattern recognition receptors. Interestingly, cytosolic exposure of double-stranded RNA was delayed in porcine cells compared to primate cells, independent of total double-stranded RNA levels and in correlation with the timing of the interferon response. Furthermore, when double-stranded RNA was artificially introduced into the cytosol of porcine cells, more rapid and robust interferon activation was triggered than in viral infection. Thus, cytosolic exposure of JEV double-stranded RNA is imperative for interferon induction, but in cell lines (e.g., porcine cells) with delayed emergence of cytosolic double-stranded RNA, the interferon response is late and viral dissemination is consequently enhanced.
Figures

References
-
- Chang T. H., Liao C. L., Lin Y. L. 2006. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I dependent IRF-3 and PI3K-dependent NF-κB activation. Microb. Infect. 8:157–171 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
