

doi: 10.2337/db10-1797.
How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited
Affiliations
- PMID: 21525508
- PMCID: PMC3292309
- DOI: 10.2337/db10-1797
Item in Clipboard
How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited
Diabetes.
2011 May.
No abstract available
Figures
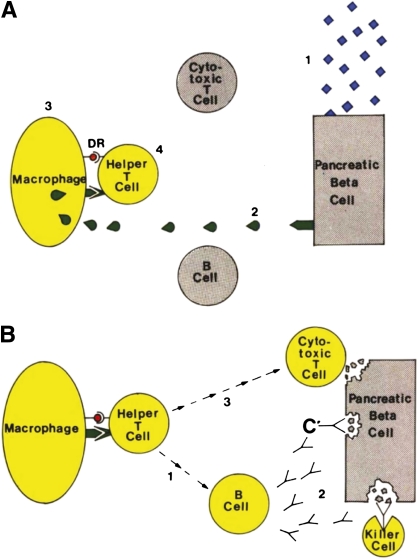
Bottazzo’s “Exhibit 5.” Diagram shows the hypothetical steps leading to activation of the immune system against the β-cell. A: The triggering events: 1) Environmental attack. 2) Release of autoantigens from the β-cell. 3) The macrophage processes them on its surface membrane. D/DR molecules present islet autoantigens to the helper T-cell. 4) Activation of the helper T-cell. B: Closing the circle: 1) Activation of the B-cell to the helper T-cell. 2) Production of islet cell antibodies, followed by antibody-dependent complement (C’) and killer cell–mediated cytotoxicity. 3) Activation of the cytotoxic T-cell. Adapted (direct prose) from Bottazzo (3), reprinted with permission from John Wiley & Sons, Inc.
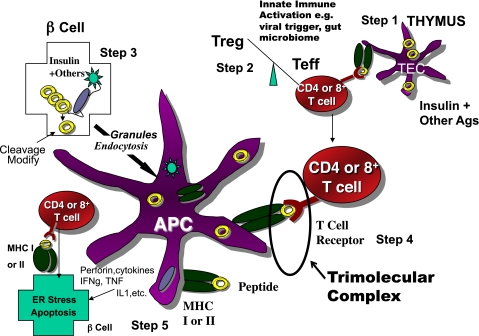
Simplified model of the immune pathogenesis of type 1 diabetes. Major components include Step 1) The thymus where peptides of peripheral antigens are expressed and presented by HLA molecules on the surface of medullary thymic epithelial cells (mTECs cells) to T-cell receptors, leading to deletion of many but not all anti-islet autoreactive T-cells. Step 2) Regulatory T-cells (Treg) and effector T-cells are both produced, and their balance is crucial for maintaining tolerance. Innate immune activation can affect the balance in terms of activating autoimmunity. Step 3) The β-cell itself not only produces target antigens but also modifies molecules, such as chromogranin, by cleavage at critical sites, thus creating peptides recognized by pathogenic T-cells. There is evidence that processing of molecules such as insulin within the β-cell creates peptides that are then taken up by antigen-presenting cells either as whole, dead β-cells, or specifically, granules of β-cells, for eventual further processing and presentation of islet peptides to effector T-cells. Step 4) The trimolecular complex, involving the MHC-presenting molecule/peptide in the appropriate “register”/T-cell receptor recognizing both and, like a lock and key, is the essential recognition unit for adaptive organ-specific autoimmunity. Step 5) Finally, CD4 T-cells orchestrate multiple arms of the immune system (e.g., CD8 cytotoxic T-cells, pathogenic cytokine production), resulting in specific destruction of islet β-cells.
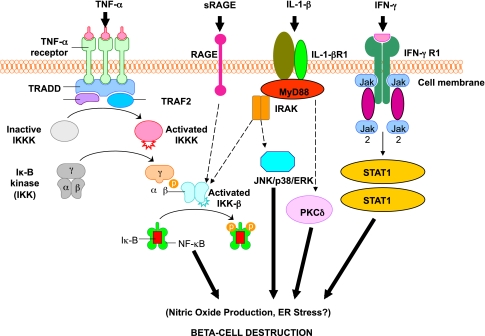
Activation of inflammatory mediators in pancreatic β-cells in type 1 diabetes. Tumor necrosis factor-α (TNF-α), IL-1β, and interferon-γ (IFN-γ) are the most likely cytokines acting in synergy during inflammation of pancreatic β-cells, leading to the activation of a final common pathway, such as nuclear factor-κB (NF-κB) and, ultimately, to β-cell destruction. NF-κB can be activated by a variety of stimuli, including TNF-α, IL-1, receptor for advanced glycation end products (RAGE), and Toll-like receptors (TLRs). IL-1β is an inflammatory cytokine that plays a major role in immune-mediated β-cell destruction. Interestingly, in patients with type 2 diabetes, the IL-1 pathway blockade with an IL-1 receptor antagonist (Anakinra) improved glycemic control and β-cell secretory function and resulted in a significant reduction marker of systemic inflammation, namely, C-reactive protein and IL-6 (56). A recent clinical study indicated that the blockade of the IL-1β pathway in type 1 diabetes resulted in the reduced ability of mononuclear cells to traffic to sites of inflammation (57). The latter observations provide evidence for a possible mechanistic link between type 1 and type 2 diabetes, and additional studies are necessary to unravel the common inflammatory pathways involved in the pathologic etiology of these two diseases. Compelling evidence indicates that cytokines influence the expression of inducible NO synthase (iNOS) leading to NO production. IL-1β and IFNγ, by NO synthesis, were reported to markedly decrease sarco(endo)plasmic reticulum Ca2+ ATPase 2b (SERCA2b) protein expression, deplete Ca2+ stores, and activate ER stress pathway, which is a potential contributing mechanism to β-cell death. Furthermore, cytokine-induced (IL-1β + IFN-γ) apoptosis of INS-1 cells appears to depend on NO production, as demonstrated by the use of the NO dioxygenase blocker NG-methyl-l -arginine. NO also contributes to cytokine-induced apoptosis through potentiation of Jun NH2-terminal kinase (JNK) activity and suppression of Akt/protein kinase B. Although whether oxidative stress plays a key role in the pathogenesis of type 1 diabetes is still being discussed, a reduced antioxidant capacity has been demonstrated in patients with type 1 diabetes compared with healthy control subjects. To summarize the cytokine signaling, TNF-α signals through trimerized p60 receptors that interact with the TNF receptor type 1–associated death domain protein (TRADD). Fas-associated protein with death domain (FADD) is then recruited by TRADD, thus allowing binding of receptor-interacting protein (RIP) and TNF receptor–associated factor 2 (TRAF2) to the receptor complex. TRAF2 activates NF-κB through NF-κB–inducing kinase (NIK)–inhibitor of κB kinase (IKK) and activates the JNK/p38 pathways. TNF-α is an inflammatory cytokine that appears to be associated with a number of autoimmune disorders, including type 1 diabetes. TNF-α may activate intraislet resident macrophages, resulting in the release of IL-1β, which generates iNOS expression and the overproduction of NO in β-cells. Alterations in the number and function of CD4+CD25+ T-cells may be an additional mechanism by which TNF-α may cause type 1 diabetes in NOD mice. The role of RAGE mediated by NF-κB has not been entirely elucidated, although RAGE may be an important intermediary in causing monocyte production of inflammatory mediators such as TNF-α. It is possible that increased expression of RAGE in response to hyperglycemia may lead to activation of innate and even adaptive immune responses and enhance β-cell destruction. After IL-1β binding to IL-1βR1, MyD88 is recruited to the receptor complex. MyD88 interacts with IL-1 receptor–associated kinase (IRAK), allowing the binding of TRAF6 to IRAK. TRAF6 causes activation of mitogen-activated protein kinase/stress-activated protein kinase and activation of the NF-κB pathway by transforming growth factor-β–activated kinase 1 (TAK1)–mediated activation of IKK. IL-1β also stimulates activation of protein kinase C-δ (PKC-δ), possibly through phospholipase C generation of diacylglycerol. ERK, extracellular signal–regulated kinase; Jak, Janus kinase; STAT1, signal transducer and activator of transcription-1.
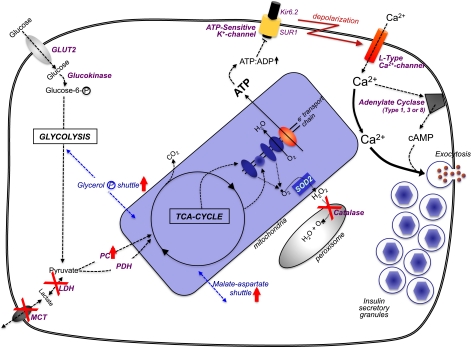
The pancreatic β-cell’s “metabolic wiring” is uniquely geared to generate secondary signals. Glucose enters the β-cell by the GLUT2 transporter, where it is phosphorylated by glucokinase and channeled to glycolysis. The combination of GLUT2 and glucokinase (with their Km values in the mmol/L range) sense glucose in the physiologic range that is unique to only a few nutrient-sensing cells in the body. Also relatively unique to β-cells is a negligible amount of lactate dehydrogenase (LDH) and the plasma membrane lactate/pyruvate transporter (MCT), so that the pyruvate resulting from glycolysis is channeled to the trichloroacetic acid cycle in the mitochondria. To counter for this, there is unusually high pyruvate carboxylase (PC) activity in β-cells to accompany pyruvate dehydrogenase (PDH). To rebalance β-cell redox in the absence of LDH/MCT, there is an increase in mitochondrial shuttle activities, particularly the glycerol-phosphate and malate-aspartate shuttles. Mitochondrial oxidative activity by the trichloroacetic acid cycle and electron transport chain generates [ATP] that is comparable to the glycolytic flux relative to increased circulating glucose. A byproduct of increased mitochondrial oxidative activity is a rise in oxygen radicals that can be converted to H2O2 in mitochondria by superoxide dismutase-2 (SOD2). But β-cells cannot dispose of H2O2 very well because they are uniquely deficient in catalase in their peroxisomes. An increase in ATP production in β-cells leads to a rise in the adenosine 5´-triphosphate (ATP)/adenosine 5´-diphosphate (ADP) ratio that shuts the ATP-sensitive potassium channel, which consists of the sulfonylurea receptor (SUR1) and Kir6.2 subunits. The consequential decreased efflux of K+ results in a depolarization of the β-cell’s plasma membrane, and voltage-sensitive Ca2+-channels open, increasing cytosolic [Ca2+]i. The rise in [Ca2+]i is the major trigger to induce insulin secretory granule exocytosis for release of insulin to the circulation. The rise in [Ca2+]i can also activate Ca2+/calmodulin–activated adenylate cyclases (type I, II, or VIII) to increase the cell’s [cAMP]i levels, which is an augmentation signal to potentiate the Ca2+-induced insulin secretion.
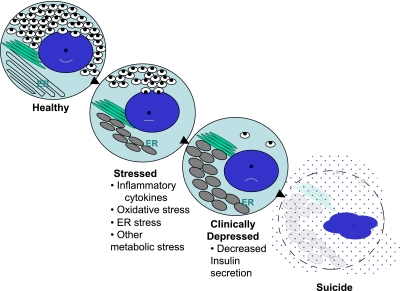
The progressive state of β-cell dysfunction in type 1 diabetes—a model for stages of depression? As noted in the text, increasing data support a model of progressive β-cell dysfunction in type 1 diabetes. Meaning, from the activities of a variety of “stressors,” (e.g., inflammation, glucolipotoxicity, ER stress, etc.), β-cells move from a state of normalcy to a “stressed” state by a process that depletes the insulin storage pool. Therefore, β-cells proceed to a state where dysfunctional insulin secretion occurs, along with expansion and distortion of the ER. The end stage is one of “assisted suicide,” where the immunologic parameters, when combined with unique susceptibilities inherent to β-cells, lead to their ultimate demise.
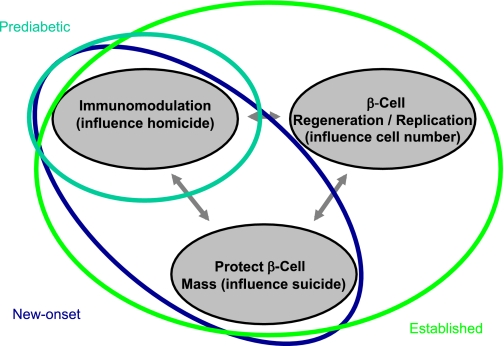
An idealized model illustrates intervention in type 1 diabetes by the degree of disease state and improved knowledge of understanding the role of β-cell homicide (i.e., immune-mediated death) and suicide (i.e., mechanisms that contribute to cellular death) in the processes underlying the formation of this disorder. Subjects would, theoretically, be placed into different means and methods of therapy by the status of their disease state, such as prediabetic, new-onset diabetes, or established diabetes. On the basis of our understanding of the natural history of diabetes, an increasingly intensive intervention may be required to provide therapeutic benefit to those with this disease. For example, in those with ongoing anti–β-cell autoimmunity but who lack overt type 1 diabetes (aqua line), therapy with a single agent capable of restoring immune tolerance and disrupting immune-mediated death may be beneficial. However, for patients with new-onset disease (blue line), combination therapy that provides an agent capable of restoring immune tolerance along with a drug that preserves and protects the remaining β-cells may prove ideal. Finally, for the patient with established (i.e., long-term) type 1 diabetes (green line), a three-way combination therapy that includes the aforementioned agent forms with a therapy capable of inducing β-cell replication/neogenesis may prove most beneficial.
References
-
- Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 2001;358:221–229 - PubMed
-
- Bottazzo GF. Lawrence lecture. Death of a beta cell: homicide or suicide? Diabet Med 1986;3:119–130 - PubMed
-
- Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis 2009;14:1389–1404 - PubMed
-
- Seewaldt S, Thomas HE, Ejrnaes M, et al. Virus-induced autoimmune diabetes: most beta-cells die through inflammatory cytokines and not perforin from autoreactive (anti-viral) cytotoxic T-lymphocytes. Diabetes 2000;49:1801–1809 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
