

Proteinortho: detection of (co-)orthologs in large-scale analysis
- PMID: 21526987
- PMCID: PMC3114741
- DOI: 10.1186/1471-2105-12-124
Proteinortho: detection of (co-)orthologs in large-scale analysis
Abstract
Background: Orthology analysis is an important part of data analysis in many areas of bioinformatics such as comparative genomics and molecular phylogenetics. The ever-increasing flood of sequence data, and hence the rapidly increasing number of genomes that can be compared simultaneously, calls for efficient software tools as brute-force approaches with quadratic memory requirements become infeasible in practise. The rapid pace at which new data become available, furthermore, makes it desirable to compute genome-wide orthology relations for a given dataset rather than relying on relations listed in databases.
Results: The program Proteinortho described here is a stand-alone tool that is geared towards large datasets and makes use of distributed computing techniques when run on multi-core hardware. It implements an extended version of the reciprocal best alignment heuristic. We apply Proteinortho to compute orthologous proteins in the complete set of all 717 eubacterial genomes available at NCBI at the beginning of 2009. We identified thirty proteins present in 99% of all bacterial proteomes.
Conclusions: Proteinortho significantly reduces the required amount of memory for orthology analysis compared to existing tools, allowing such computations to be performed on off-the-shelf hardware.
Figures


 , and y1 and
, and y1 and  , resp., it is possible that there are no reciprocal best
, resp., it is possible that there are no reciprocal best 

 with the
with the  in splitting in particular large groups. While these groups are left intact for
in splitting in particular large groups. While these groups are left intact for  , thresholds of 0.5 and higher drastically reduce the fraction of included co-orthologs.
, thresholds of 0.5 and higher drastically reduce the fraction of included co-orthologs.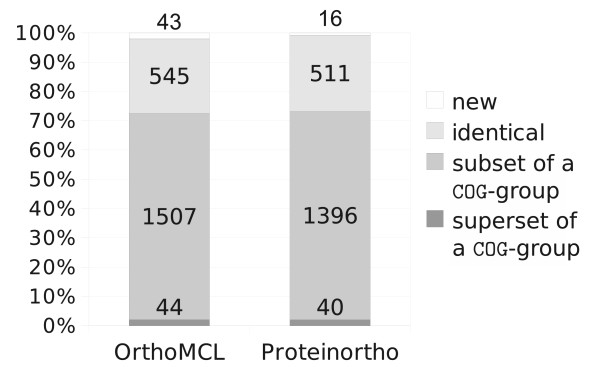

References
-
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008. pp. D13–21. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
