

Macrophages in the pathogenesis of atherosclerosis
- PMID: 21529710
- PMCID: PMC3111065
- DOI: 10.1016/j.cell.2011.04.005
Macrophages in the pathogenesis of atherosclerosis
Abstract
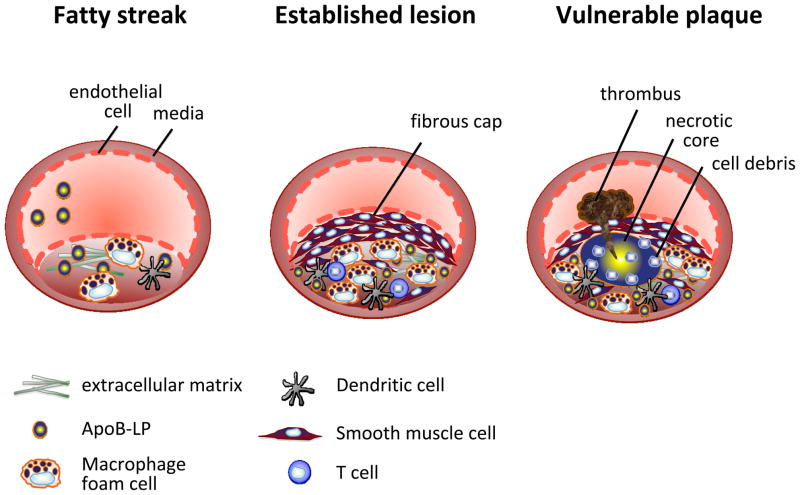
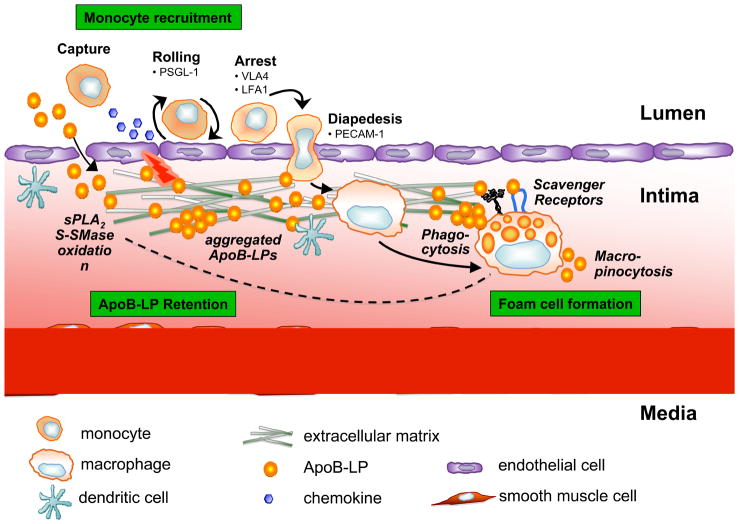
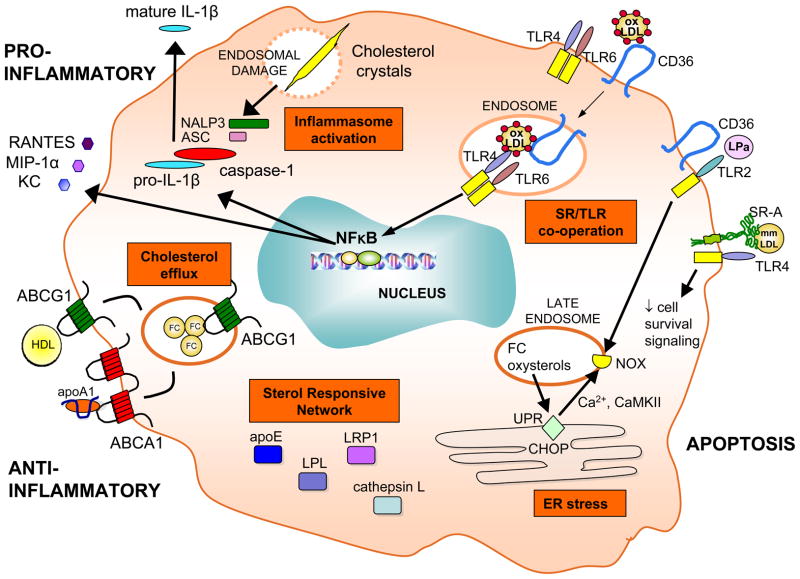
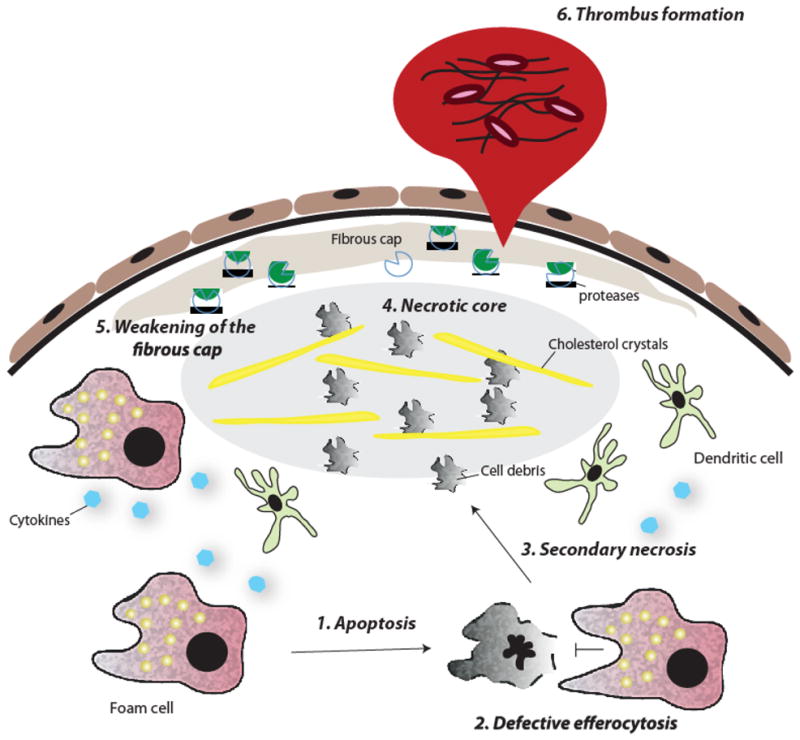
In atherosclerosis, the accumulation of apolipoprotein B-lipoproteins in the matrix beneath the endothelial cell layer of blood vessels leads to the recruitment of monocytes, the cells of the immune system that give rise to macrophages and dendritic cells. Macrophages derived from these recruited monocytes participate in a maladaptive, nonresolving inflammatory response that expands the subendothelial layer due to the accumulation of cells, lipid, and matrix. Some lesions subsequently form a necrotic core, triggering acute thrombotic vascular disease, including myocardial infarction, stroke, and sudden cardiac death. This Review discusses the central roles of macrophages in each of these stages of disease pathogenesis.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Abi-Younes S, Sauty A, Mach F, Sukhova GK, Libby P, Luster AD. The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ Res. 2000;86:131–138. - PubMed
-
- Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, Mak PA, Edwards PA, Mangelsdorf DJ, Tontonoz P, Miyazaki T. A role for the apoptosis inhibitory factor AIM/Spα/Api6 in atherosclerosis development. Cell Metabolism. 2005;1:201–213. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
