

Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]
- PMID: 21529751
- PMCID: PMC3146728
- DOI: 10.1016/j.ajhg.2011.04.003
Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]
Erratum in
- Am J Hum Genet. 2011 May 13;88(5):677
Abstract
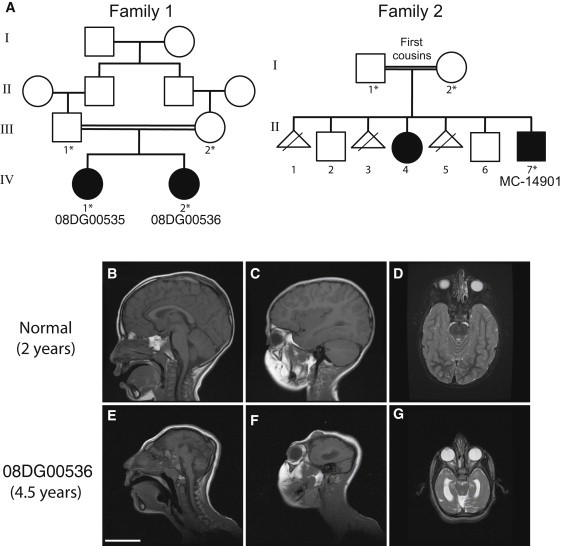
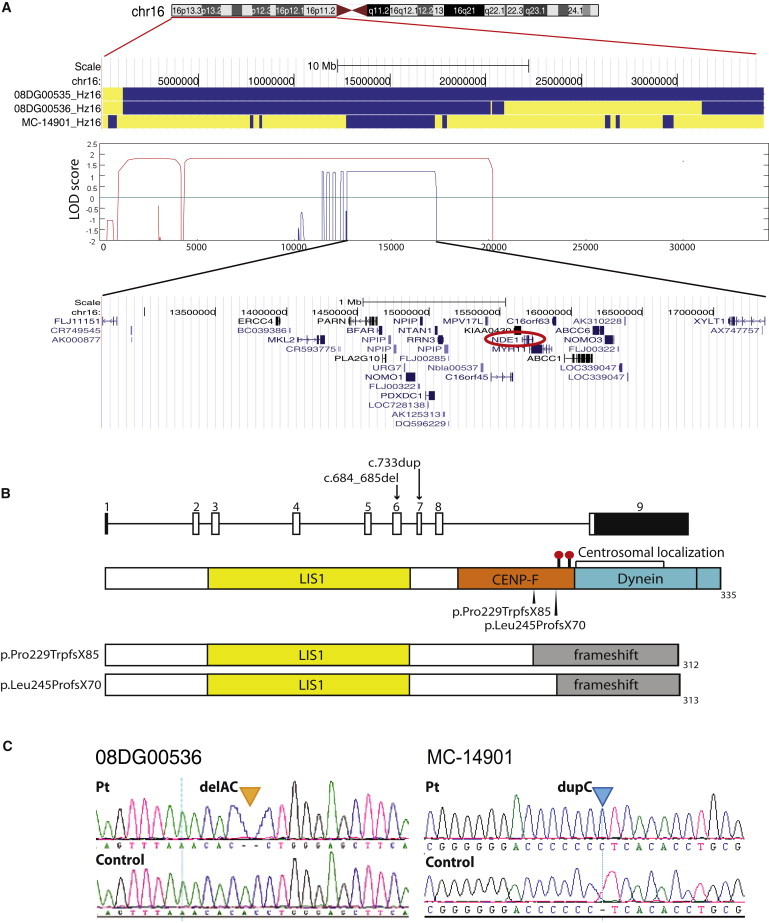
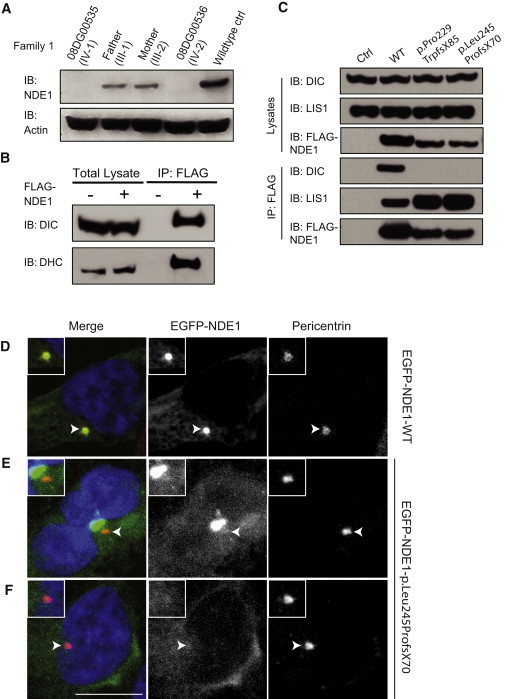
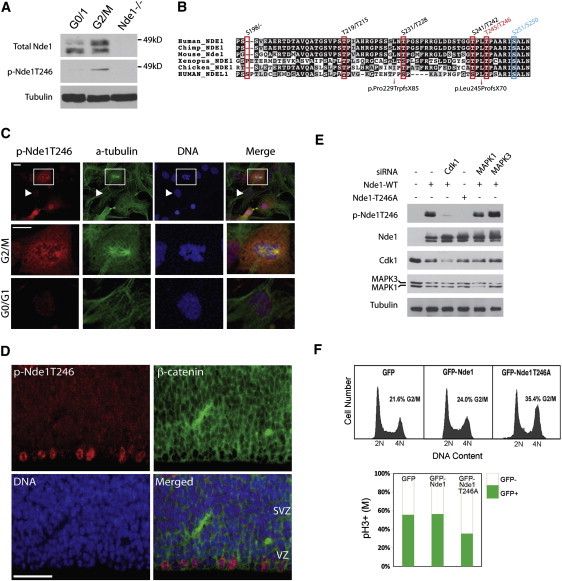
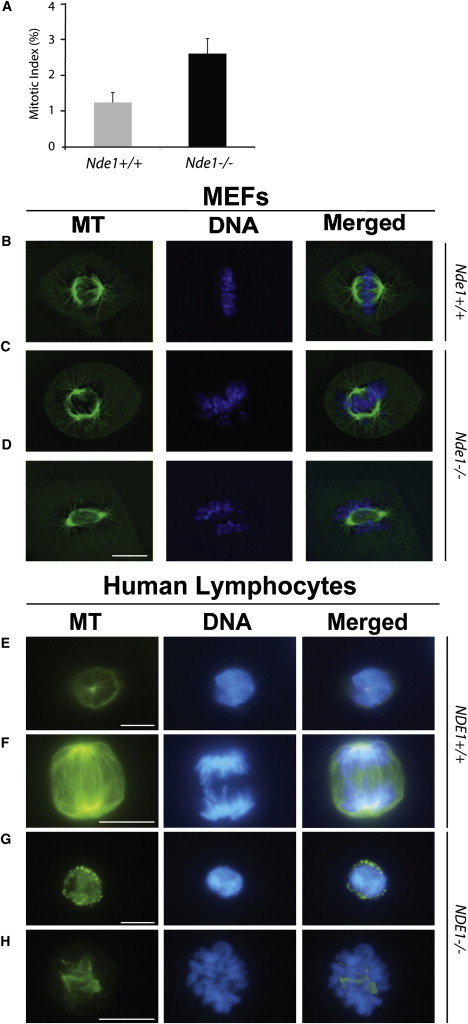
Genes disrupted in human microcephaly (meaning "small brain") define key regulators of neural progenitor proliferation and cell-fate specification. In comparison, genes mutated in human lissencephaly (lissos means smooth and cephalos means brain) highlight critical regulators of neuronal migration. Here, we report two families with extreme microcephaly and grossly simplified cortical gyral structure, a condition referred to as microlissencephaly, and show that they carry homozygous frameshift mutations in NDE1, which encodes a multidomain protein that localizes to the centrosome and mitotic spindle poles. Both human mutations in NDE1 truncate the C-terminal NDE1domains, which are essential for interactions with cytoplasmic dynein and thus for regulation of cytoskeletal dynamics in mitosis and for cell-cycle-dependent phosphorylation of NDE1 by Cdk1. We show that the patient NDE1 proteins are unstable, cannot bind cytoplasmic dynein, and do not localize properly to the centrosome. Additionally, we show that CDK1 phosphorylation at T246, which is within the C-terminal region disrupted by the mutations, is required for cell-cycle progression from the G2 to the M phase. The role of NDE1 in cell-cycle progression probably contributes to the profound neuronal proliferation defects evident in Nde1-null mice and patients with NDE1 mutations, demonstrating the essential role of NDE1 in human cerebral cortical neurogenesis.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Comment in
-
The essential role of NDE1 in extreme microcephaly.Clin Genet. 2011 Sep;80(3):241-2. doi: 10.1111/j.1399-0004.2011.01753.x. Clin Genet. 2011. PMID: 21762443 No abstract available.
References
-
- Dobyns W.B., Reiner O., Carrozzo R., Ledbetter D.H. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270:2838–2842. - PubMed
-
- Gleeson J.G., Allen K.M., Fox J.W., Lamperti E.D., Berkovic S., Scheffer I., Cooper E.C., Dobyns W.B., Minnerath S.R., Ross M.E., Walsh C.A. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. - PubMed
-
- Hong S.E., Shugart Y.Y., Huang D.T., Shahwan S.A., Grant P.E., Hourihane J.O., Martin N.D., Walsh C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000;26:93–96. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 NS032457/NS/NINDS NIH HHS/United States
- R01 NS 32457/NS/NINDS NIH HHS/United States
- N01 HG065403/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 NS035129/NS/NINDS NIH HHS/United States
- R01HD56380/HD/NICHD NIH HHS/United States
- P30 HD18655/HD/NICHD NIH HHS/United States
- R21 NS061772/NS/NINDS NIH HHS/United States
- T32 GM007726-35/GM/NIGMS NIH HHS/United States
- P30 HD018655/HD/NICHD NIH HHS/United States
- HHSN268200782096C/HG/NHGRI NIH HHS/United States
- R01 HD056380/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
