

The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation
- PMID: 21531565
- PMCID: PMC3112285
- DOI: 10.1016/j.tibs.2011.03.006
The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation
Abstract
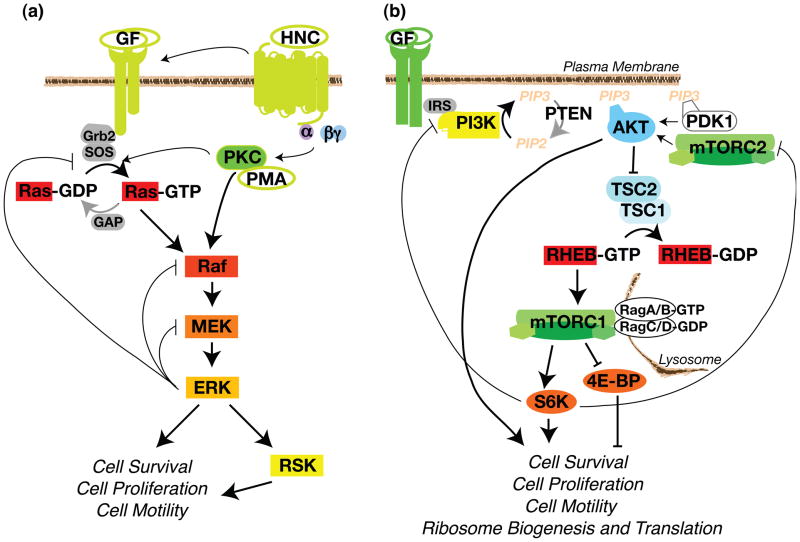
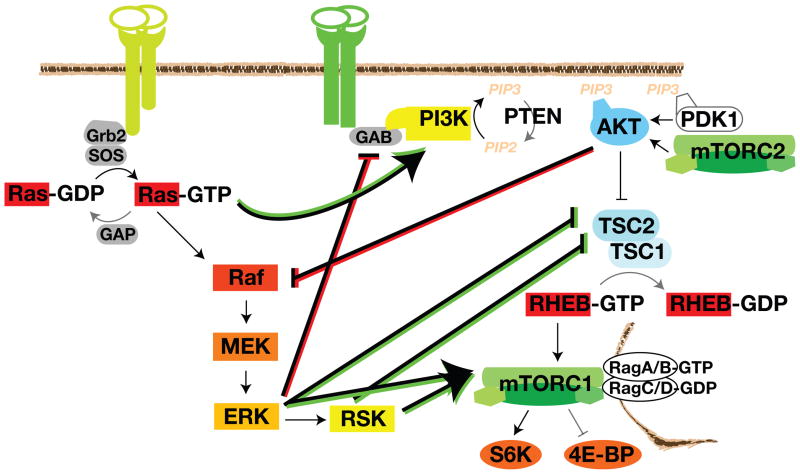
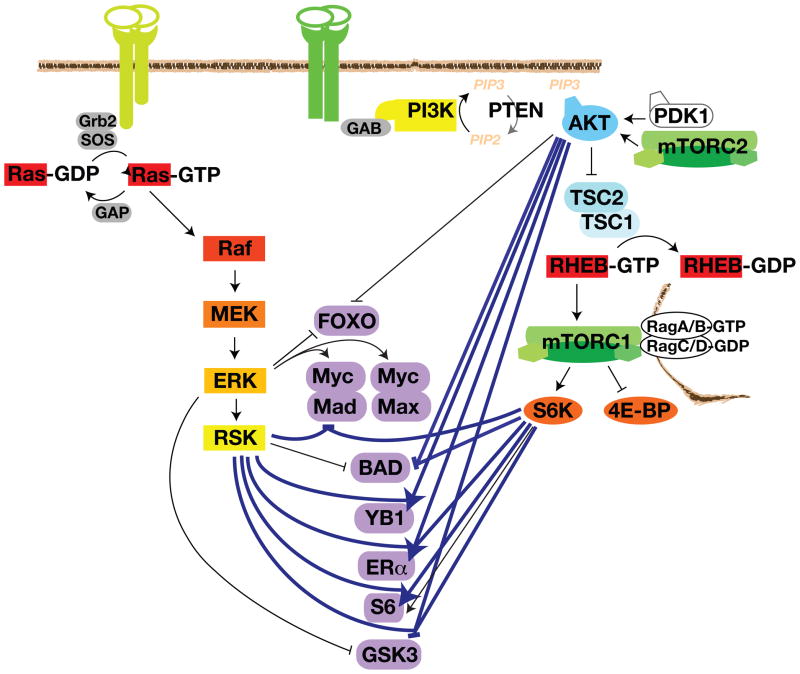
The Ras-extracellular signal-regulated kinase (Ras-ERK) and phosphatidylinositol 3-kinase-mammalian target of rapamycin (PI3K-mTOR) signaling pathways are the chief mechanisms for controlling cell survival, differentiation, proliferation, metabolism, and motility in response to extracellular cues. Components of these pathways were among the first to be discovered when scientists began cloning proto-oncogenes and purifying cellular kinase activities in the 1980s. Ras-ERK and PI3K-mTOR were originally modeled as linear signaling conduits activated by different stimuli, yet even early experiments hinted that they might intersect to regulate each other and co-regulate downstream functions. The extent of this cross-talk and its significance in cancer therapeutics are now becoming clear.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
References
-
- McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26(22):3113–21. - PubMed
-
- Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213(3):589–602. - PubMed
-
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9(10):747–58. - PubMed
-
- Dhillon AS, et al. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–90. - PubMed
-
- Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
