

Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome
- PMID: 21533187
- PMCID: PMC3077396
- DOI: 10.1371/journal.pgen.1002050
Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome
Abstract
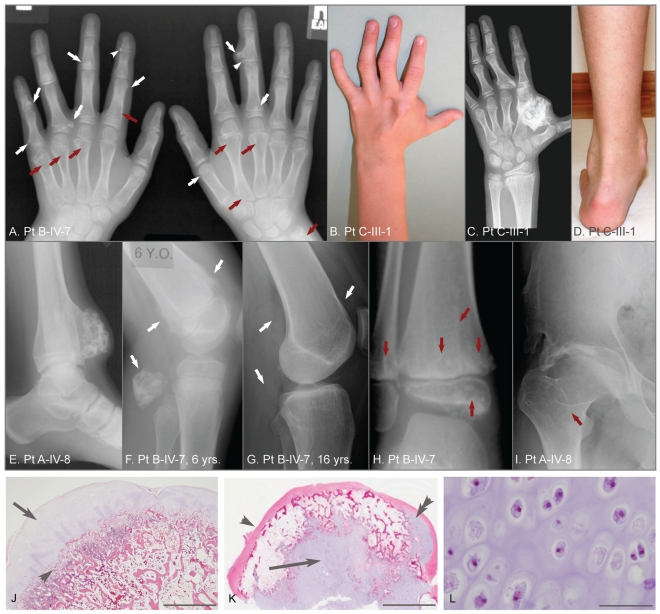
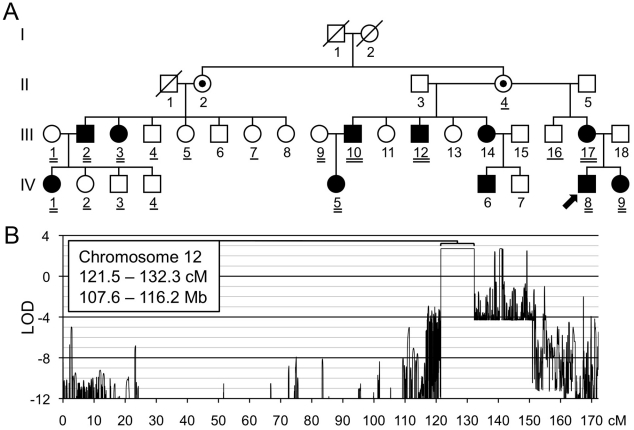
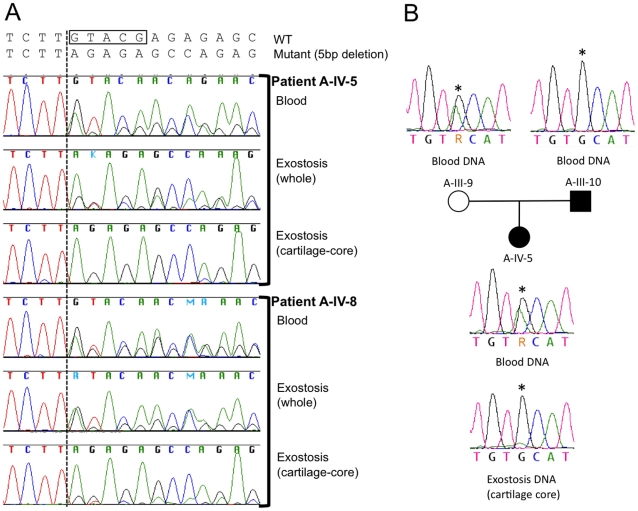
Metachondromatosis (MC) is a rare, autosomal dominant, incompletely penetrant combined exostosis and enchondromatosis tumor syndrome. MC is clinically distinct from other multiple exostosis or multiple enchondromatosis syndromes and is unlinked to EXT1 and EXT2, the genes responsible for autosomal dominant multiple osteochondromas (MO). To identify a gene for MC, we performed linkage analysis with high-density SNP arrays in a single family, used a targeted array to capture exons and promoter sequences from the linked interval in 16 participants from 11 MC families, and sequenced the captured DNA using high-throughput parallel sequencing technologies. DNA capture and parallel sequencing identified heterozygous putative loss-of-function mutations in PTPN11 in 4 of the 11 families. Sanger sequence analysis of PTPN11 coding regions in a total of 17 MC families identified mutations in 10 of them (5 frameshift, 2 nonsense, and 3 splice-site mutations). Copy number analysis of sequencing reads from a second targeted capture that included the entire PTPN11 gene identified an additional family with a 15 kb deletion spanning exon 7 of PTPN11. Microdissected MC lesions from two patients with PTPN11 mutations demonstrated loss-of-heterozygosity for the wild-type allele. We next sequenced PTPN11 in DNA samples from 54 patients with the multiple enchondromatosis disorders Ollier disease or Maffucci syndrome, but found no coding sequence PTPN11 mutations. We conclude that heterozygous loss-of-function mutations in PTPN11 are a frequent cause of MC, that lesions in patients with MC appear to arise following a "second hit," that MC may be locus heterogeneous since 1 familial and 5 sporadically occurring cases lacked obvious disease-causing PTPN11 mutations, and that PTPN11 mutations are not a common cause of Ollier disease or Maffucci syndrome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
-
- Maroteaux P. [Metachondromatosis]. Z Kinderheilkd. 1971;109:246–261. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
