

Molecular docking: a powerful approach for structure-based drug discovery
- PMID: 21534921
- PMCID: PMC3151162
- DOI: 10.2174/157340911795677602
Molecular docking: a powerful approach for structure-based drug discovery
Abstract
Molecular docking has become an increasingly important tool for drug discovery. In this review, we present a brief introduction of the available molecular docking methods, and their development and applications in drug discovery. The relevant basic theories, including sampling algorithms and scoring functions, are summarized. The differences in and performance of available docking software are also discussed. Flexible receptor molecular docking approaches, especially those including backbone flexibility in receptors, are a challenge for available docking methods. A recently developed Local Move Monte Carlo (LMMC) based approach is introduced as a potential solution to flexible receptor docking problems. Three application examples of molecular docking approaches for drug discovery are provided.
Figures
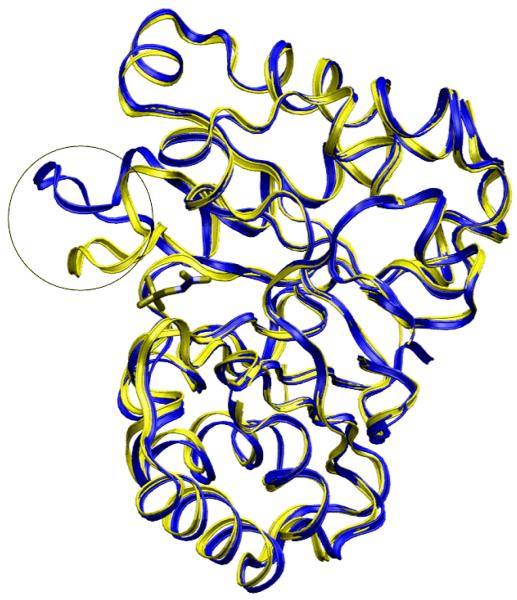
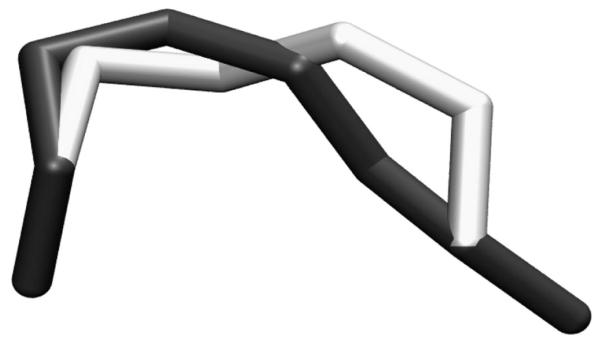

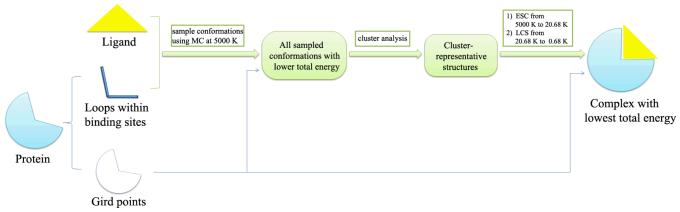
References
-
- Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303(5665):1813–1818. - PubMed
-
- Bajorath J. Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002;1(11):882–894. - PubMed
-
- Walters WP, Stahl MT, Murcko MA. Virtual screening - an overview. Drug Discov. Today. 1998;3:160–178.
-
- Langer T, Hoffmann RD. Virtual screening: an effective tool for lead structure discovery? Curr Pharm Des. 2001;7(7):509–527. - PubMed
-
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
