

HD CAG-correlated gene expression changes support a simple dominant gain of function
- PMID: 21536587
- PMCID: PMC3118763
- DOI: 10.1093/hmg/ddr195
HD CAG-correlated gene expression changes support a simple dominant gain of function
Abstract
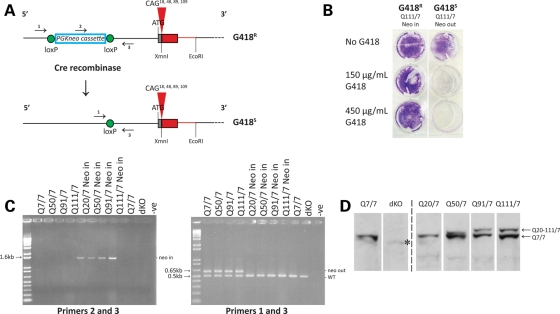
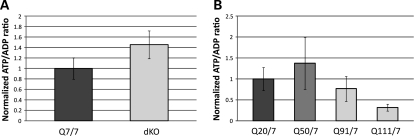
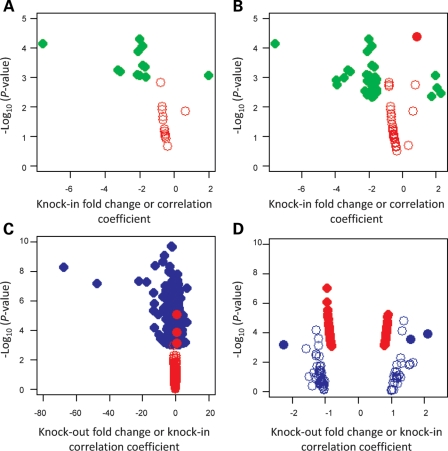
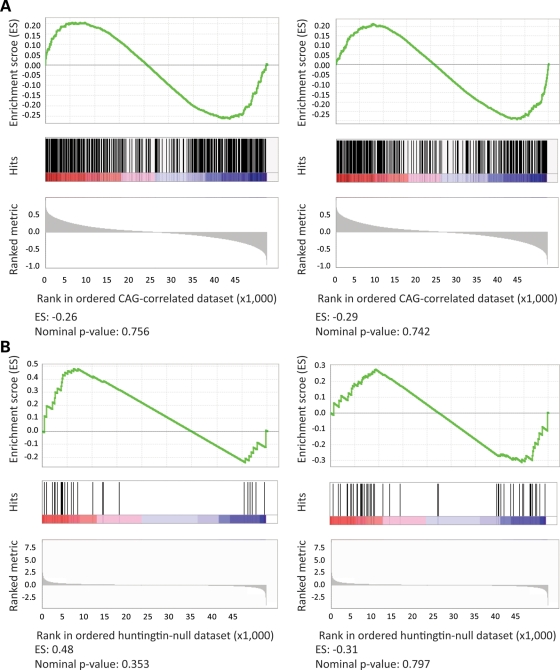
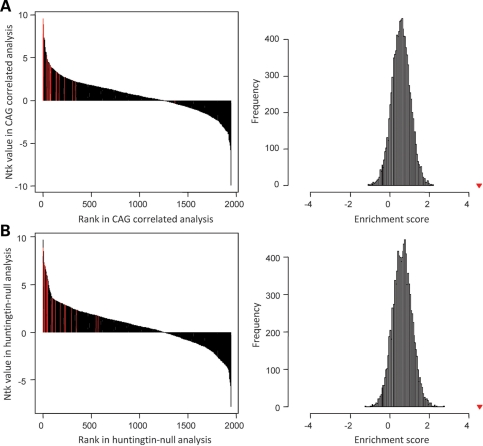
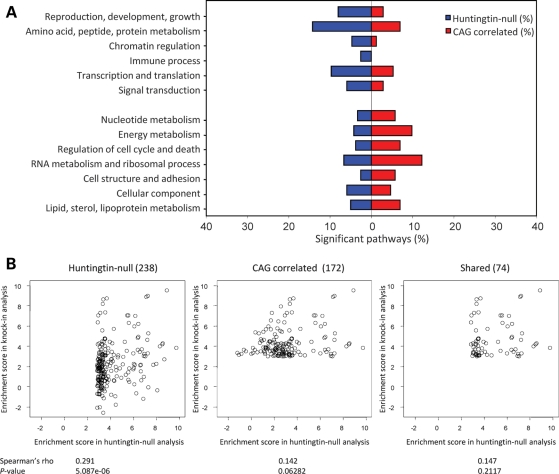
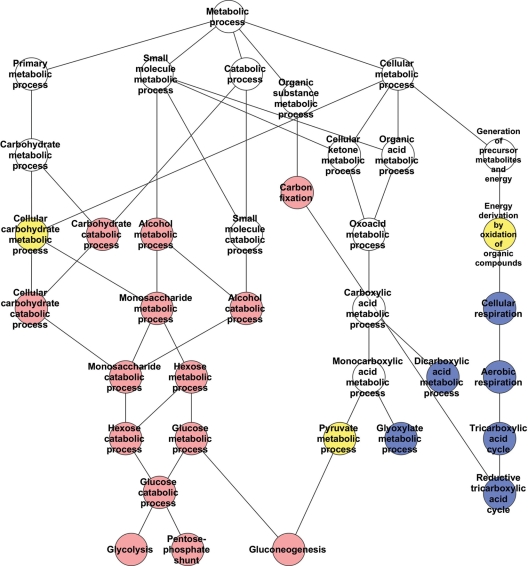
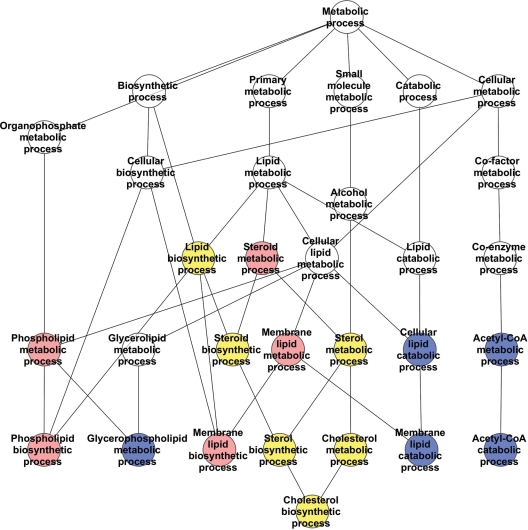
Huntington's disease is initiated by the expression of a CAG repeat-encoded polyglutamine region in full-length huntingtin, with dominant effects that vary continuously with CAG size. The mechanism could involve a simple gain of function or a more complex gain of function coupled to a loss of function (e.g. dominant negative-graded loss of function). To distinguish these alternatives, we compared genome-wide gene expression changes correlated with CAG size across an allelic series of heterozygous CAG knock-in mouse embryonic stem (ES) cell lines (Hdh(Q20/7), Hdh(Q50/7), Hdh(Q91/7), Hdh(Q111/7)), to genes differentially expressed between Hdh(ex4/5/ex4/5) huntingtin null and wild-type (Hdh(Q7/7)) parental ES cells. The set of 73 genes whose expression varied continuously with CAG length had minimal overlap with the 754-member huntingtin-null gene set but the two were not completely unconnected. Rather, the 172 CAG length-correlated pathways and 238 huntingtin-null significant pathways clustered into 13 shared categories at the network level. A closer examination of the energy metabolism and the lipid/sterol/lipoprotein metabolism categories revealed that CAG length-correlated genes and huntingtin-null-altered genes either were different members of the same pathways or were in unique, but interconnected pathways. Thus, varying the polyglutamine size in full-length huntingtin produced gene expression changes that were distinct from, but related to, the effects of lack of huntingtin. These findings support a simple gain-of-function mechanism acting through a property of the full-length huntingtin protein and point to CAG-correlative approaches to discover its effects. Moreover, for therapeutic strategies based on huntingtin suppression, our data highlight processes that may be more sensitive to the disease trigger than to decreased huntingtin levels.
Figures
References
-
- Vonsattel J.P., DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. - PubMed
-
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- McNeil S.M., Novelletto A., Srinidhi J., Barnes G., Kornbluth I., Altherr M.R., Wasmuth J.J., Gusella J.F., MacDonald M.E., Myers R.H. Reduced penetrance of the Huntington's disease mutation. Hum. Mol. Genet. 1997;6:775–779. - PubMed
-
- Kenney C., Powell S., Jankovic J. Autopsy-proven Huntington's disease with 29 trinucleotide repeats. Mov. Disord. 2007;22:127–130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
