

RNA-sequence analysis of human B-cells
- PMID: 21536721
- PMCID: PMC3106332
- DOI: 10.1101/gr.116335.110
RNA-sequence analysis of human B-cells
Abstract
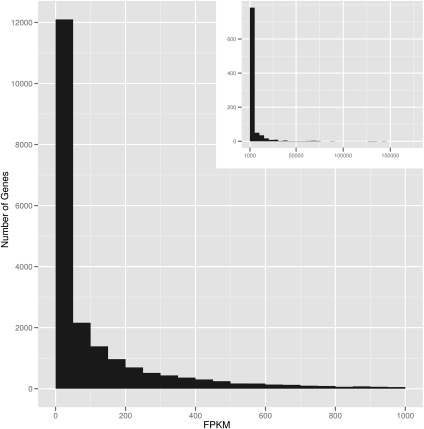
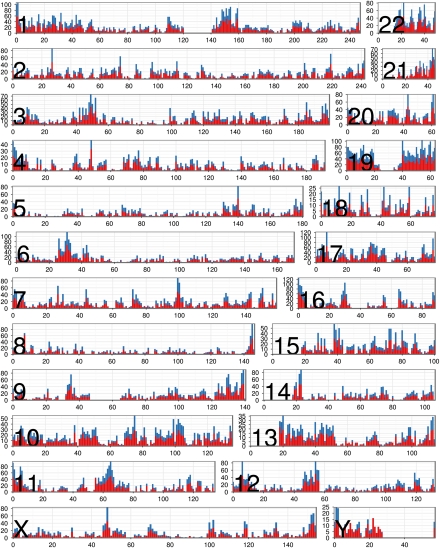
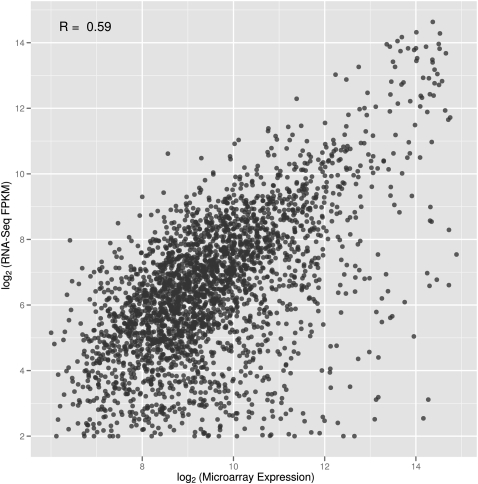
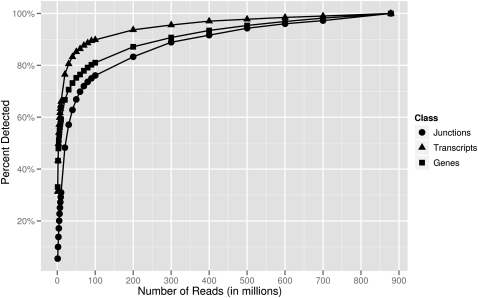
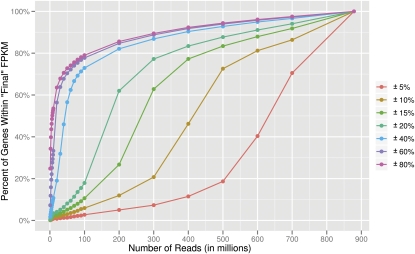
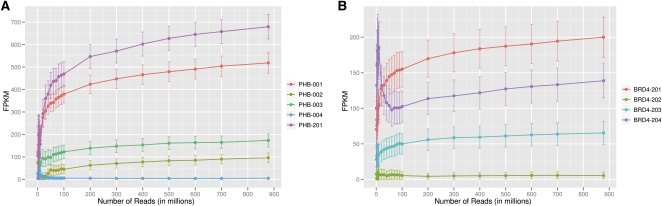
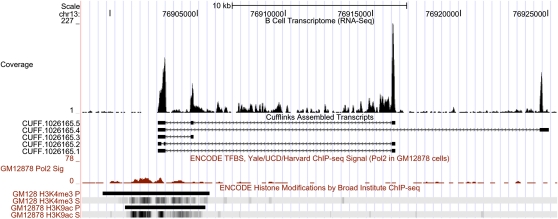
RNA-sequencing (RNA-seq) allows quantitative measurement of expression levels of genes and their transcripts. In this study, we sequenced complementary DNA fragments of cultured human B-cells and obtained 879 million 50-bp reads comprising 44 Gb of sequence. The results allowed us to study the gene expression profile of B-cells and to determine experimental parameters for sequencing-based expression studies. We identified 20,766 genes and 67,453 of their alternatively spliced transcripts. More than 90% of the genes with multiple exons are alternatively spliced; for most genes, one isoform is predominantly expressed. We found that while chromosomes differ in gene density, the percentage of transcribed genes in each chromosome is less variable. In addition, genes involved in related biological processes are expressed at more similar levels than genes with different functions. Besides characterizing gene expression, we also used the data to investigate the effect of sequencing depth on gene expression measurements. While 100 million reads are sufficient to detect most expressed genes and transcripts, about 500 million reads are needed to measure accurately their expression levels. We provide examples in which deep sequencing is needed to determine the relative abundance of genes and their isoforms. With data from 20 individuals and about 40 million sequence reads per sample, we uncovered only 21 alternatively spliced, multi-exon genes that are not in databases; this result suggests that at this sequence coverage, we can detect most of the known genes. Results from this project are available on the UCSC Genome Browser to allow readers to study the expression and structure of genes in human B-cells.
Figures
References
-
- Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D, Luo S, McCurdy S, Foy M, Ewan M, et al. 2000. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol 18: 630–634 - PubMed
-
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, et al. 2008. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5: 613–619 - PubMed
-
- Dausset J, Cann H, Cohen D, Lathrop M, Lalouel JM, White R 1990. Centre d'etude du polymorphisme humain (CEPH): Collaborative genetic mapping of the human genome. Genomics 6: 575–577 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases