

Transcript amplification from single bacterium for transcriptome analysis
- PMID: 21536723
- PMCID: PMC3106325
- DOI: 10.1101/gr.116103.110
Transcript amplification from single bacterium for transcriptome analysis
Abstract
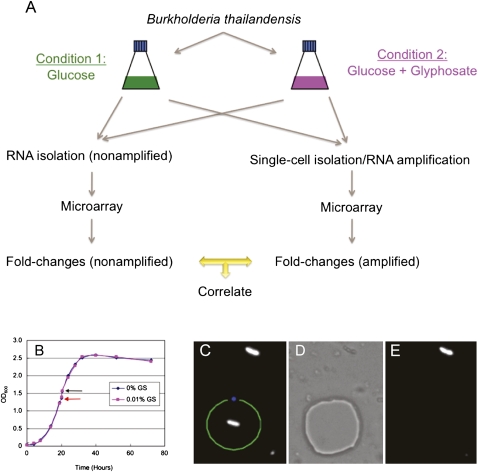
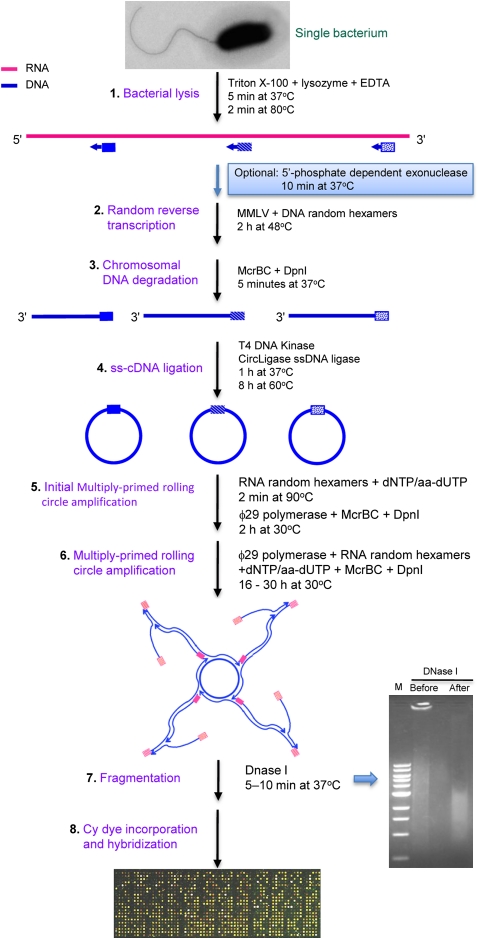
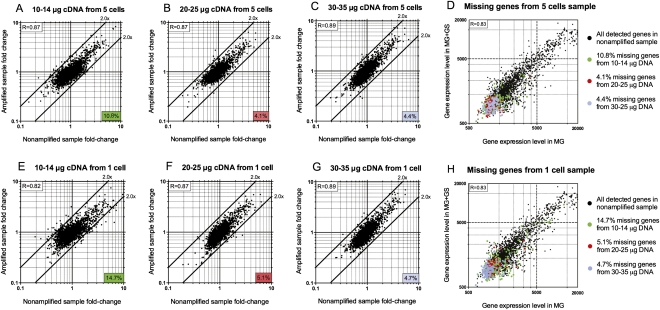
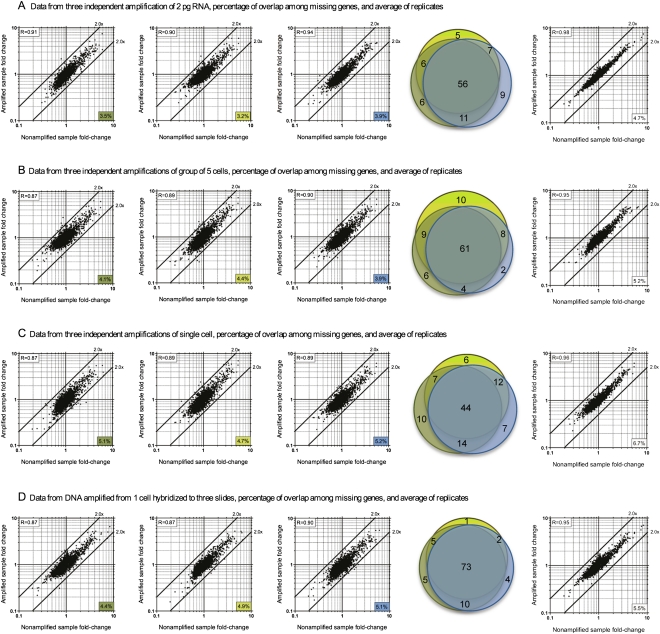
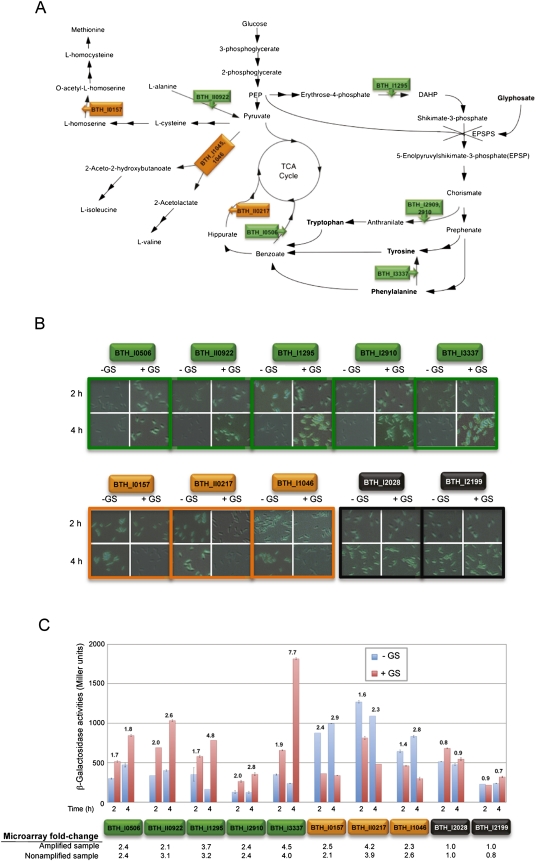
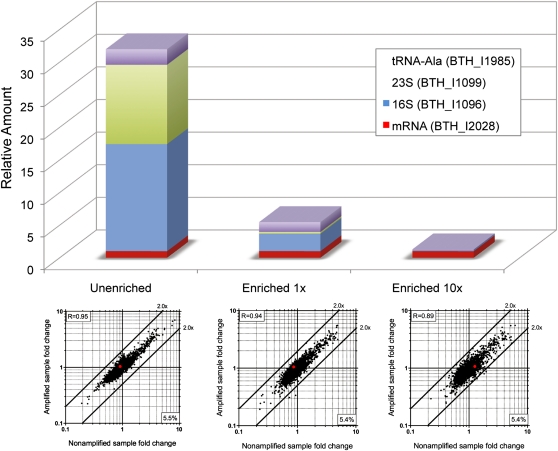
Total transcript amplification (TTA) from single eukaryotic cells for transcriptome analysis is established, but TTA from a single prokaryotic cell presents additional challenges with much less starting material, the lack of poly(A)-tails, and the fact that the messages can be polycistronic. Here, we describe a novel method for single-bacterium TTA using a model organism, Burkholderia thailandensis, exposed to a subinhibitory concentration of the antibacterial agent, glyphosate. Utilizing a B. thailandensis microarray to assess the TTA method showed low fold-change bias (less than twofold difference and Pearson correlation coefficient R ≈ 0.87-0.89) and drop-outs (4%-6% of 2842 detectable genes), compared with data obtained from the larger-scale nonamplified RNA samples. Further analysis of the microarray data suggests that B. thailandensis, when exposed to the aromatic amino acid biosynthesis inhibitor glyphosate, induces (or represses) genes to possibly recuperate and balance the intracellular amino acid pool. We validated our single-cell microarray data at the multi-cell and single-cell levels with lacZ and gfp reporter-gene fusions, respectively. Sanger sequencing of 192 clones generated from the TTA product of a single cell, with and without enrichment by elimination of rRNA and tRNA, detected only B. thailandensis sequences with no contamination. These data indicate that RNA-seq of TTA from a single cell is possible using this novel method.
Figures
References
-
- Brett PJ, DeShazer D, Woods DE 1998. Burkholderia thailandensis sp. nov., description of Burkholderia pseudomallei-like species. Int J Syst Bacteriol 48: 317–320 - PubMed
-
- Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA 1996. Laser capture microdissection. Science 274: 998–1001 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases