

WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways
- PMID: 21536751
- PMCID: PMC3087013
- DOI: 10.1083/jcb.201011051
WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways
Abstract
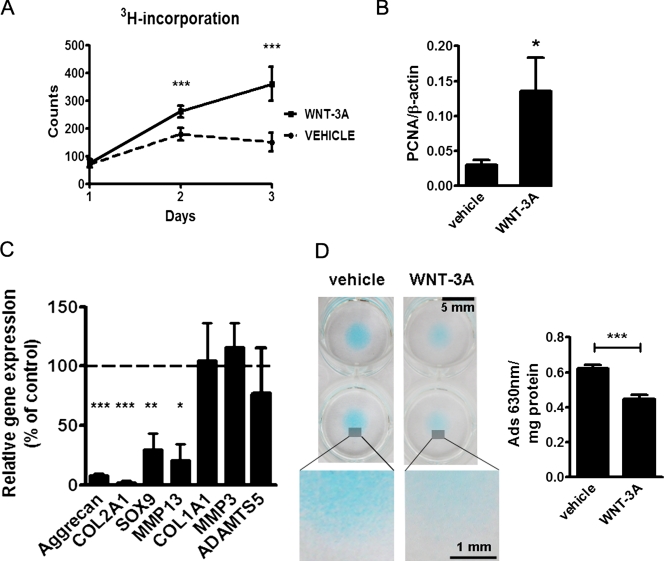
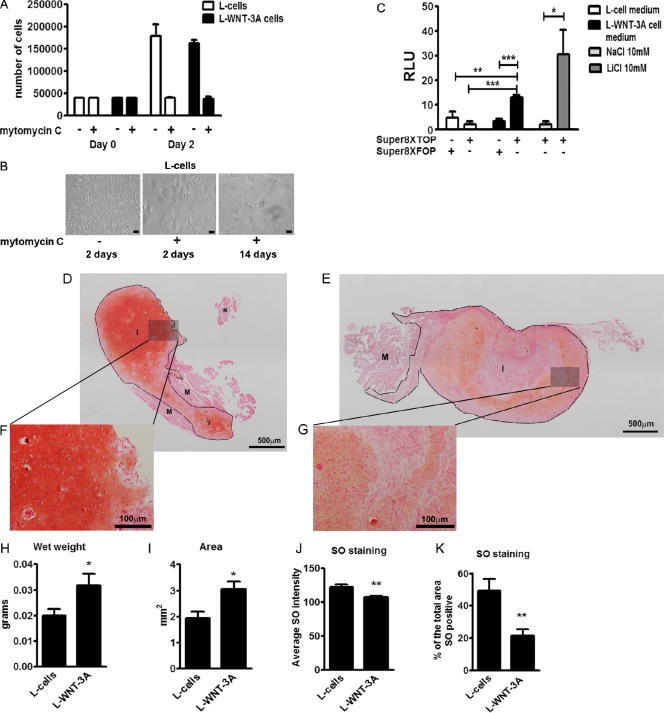
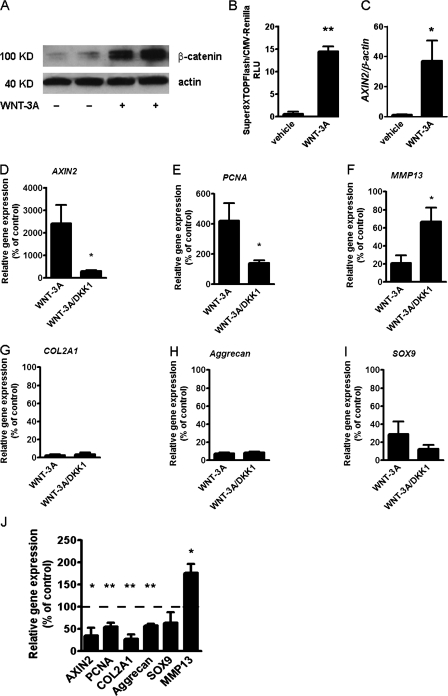
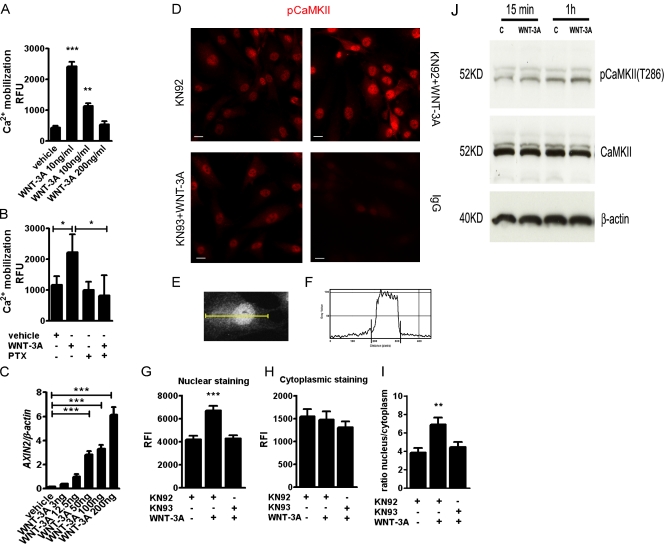
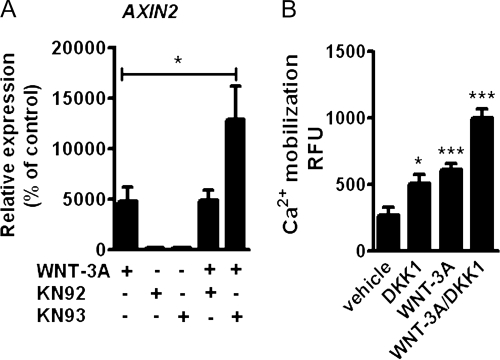
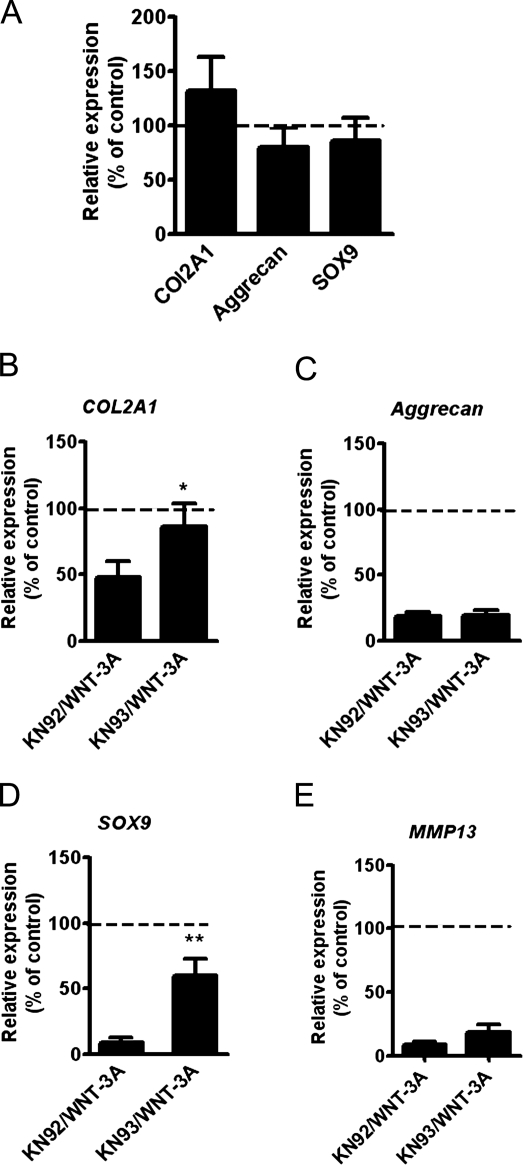
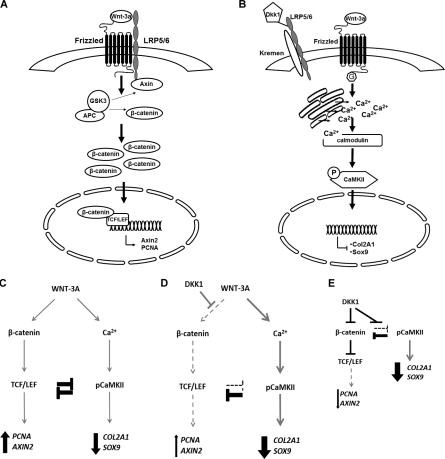
Activation and disruption of Wnt/β-catenin signaling both result in cartilage breakdown via unknown mechanisms. Here we show that both WNT-3A and the Wnt inhibitor DKK1 induced de-differentiation of human articular chondrocytes through simultaneous activation of β-catenin-dependent and independent responses. WNT-3A activates both the β-catenin-dependent canonical pathway and the Ca(2+)/CaMKII noncanonical pathways, with distinct transcriptional targets. WNT-3A promotes cell proliferation and loss of expression of the chondrocyte markers COL2A1, Aggrecan, and SOX9; however, proliferation and AXIN2 up-regulation are downstream of the canonical pathway and are rescued by DKK1, whereas the loss of differentiation markers is CaMKII dependent. Finally, we showed that in chondrocytes, the Ca(2+)/CaMKII-dependent and β-catenin-dependent pathways are reciprocally inhibitory, thereby explaining why DKK1 can induce loss of differentiation through de-repression of the CaMKII pathway. We propose a novel model in which a single WNT can simultaneously activate different pathways with distinct and independent outcomes and with reciprocal regulation. This offers an opportunity for selective pharmacological targeting.
Figures
Comment in
-
Generating a Wnt switch: it's all about the right dosage.J Cell Biol. 2011 May 2;193(3):431-3. doi: 10.1083/jcb.201103167. J Cell Biol. 2011. PMID: 21536746 Free PMC article.
References
-
- Cipolletta E., Monaco S., Maione A.S., Vitiello L., Campiglia P., Pastore L., Franchini C., Novellino E., Limongelli V., Bayer K.U., et al. 2010. Calmodulin-dependent kinase II mediates vascular smooth muscle cell proliferation and is potentiated by extracellular signal regulated kinase. Endocrinology. 151:2747–2759 10.1210/en.2009-1248 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
