

Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload
- PMID: 21537080
- PMCID: PMC3104748
- DOI: 10.1172/JCI44824
Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload
Abstract
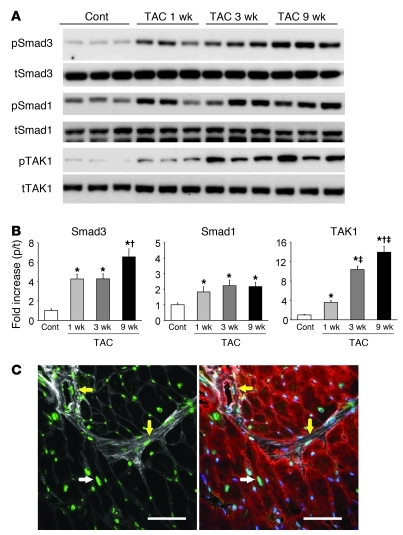
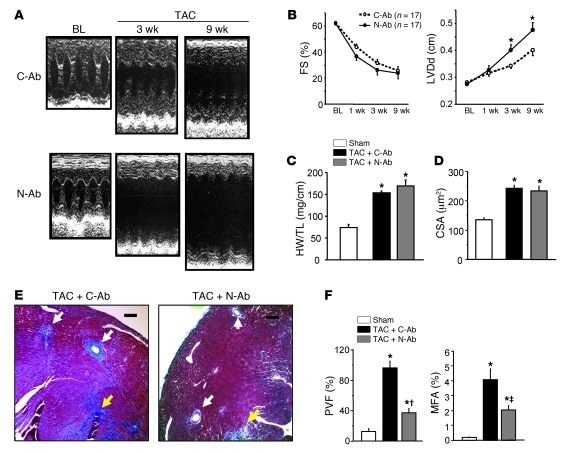
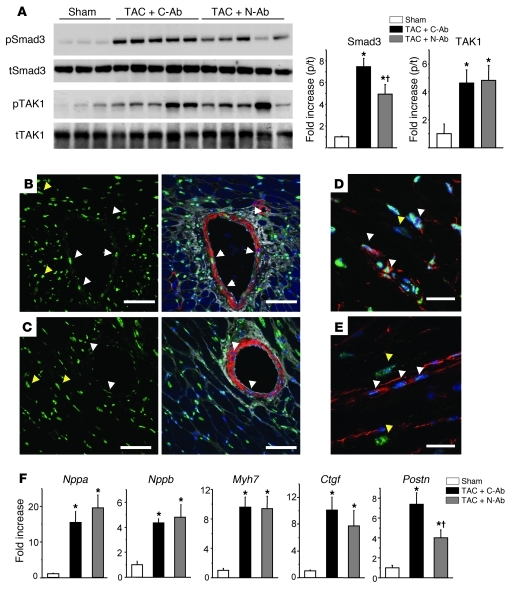
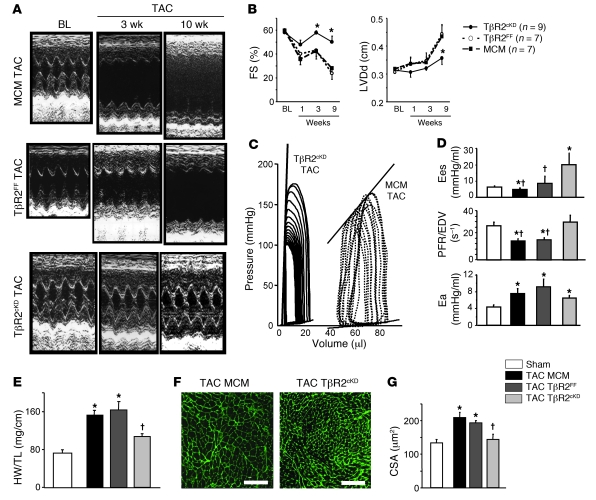
The cardiac pathological response to sustained pressure overload involves myocyte hypertrophy and dysfunction along with interstitial changes such as fibrosis and reduced capillary density. These changes are orchestrated by mechanical forces and factors secreted between cells. One such secreted factor is TGF-β, which is generated by and interacts with multiple cell types. Here we have shown that TGF-β suppression in cardiomyocytes was required to protect against maladaptive remodeling and involved noncanonical (non-Smad-related) signaling. Mouse hearts subjected to pressure overload and treated with a TGF-β-neutralizing Ab had suppressed Smad activation in the interstitium but not in myocytes, and noncanonical (TGF-β-activated kinase 1 [TAK1]) activation remained. Although fibrosis was greatly reduced, chamber dysfunction and dilation persisted. Induced myocyte knockdown of TGF-β type 2 receptor (TβR2) blocked all maladaptive responses, inhibiting myocyte and interstitial Smad and TAK1. Myocyte knockdown of TβR1 suppressed myocyte but not interstitial Smad, nor TAK1, modestly reducing fibrosis without improving chamber function or hypertrophy. Only TβR2 knockdown preserved capillary density after pressure overload, enhancing BMP7, a regulator of the endothelial-mesenchymal transition. BMP7 enhancement also was coupled to TAK1 suppression. Thus, myocyte targeting is required to modulate TGF-β in hearts subjected to pressure overload, with noncanonical pathways predominantly affecting the maladaptive hypertrophy/dysfunction.
Figures
References
-
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322(22):1561–1566. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
