

Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment
- PMID: 21538838
- PMCID: PMC3100366
- DOI: 10.1002/ajmg.a.33970
Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment
Abstract
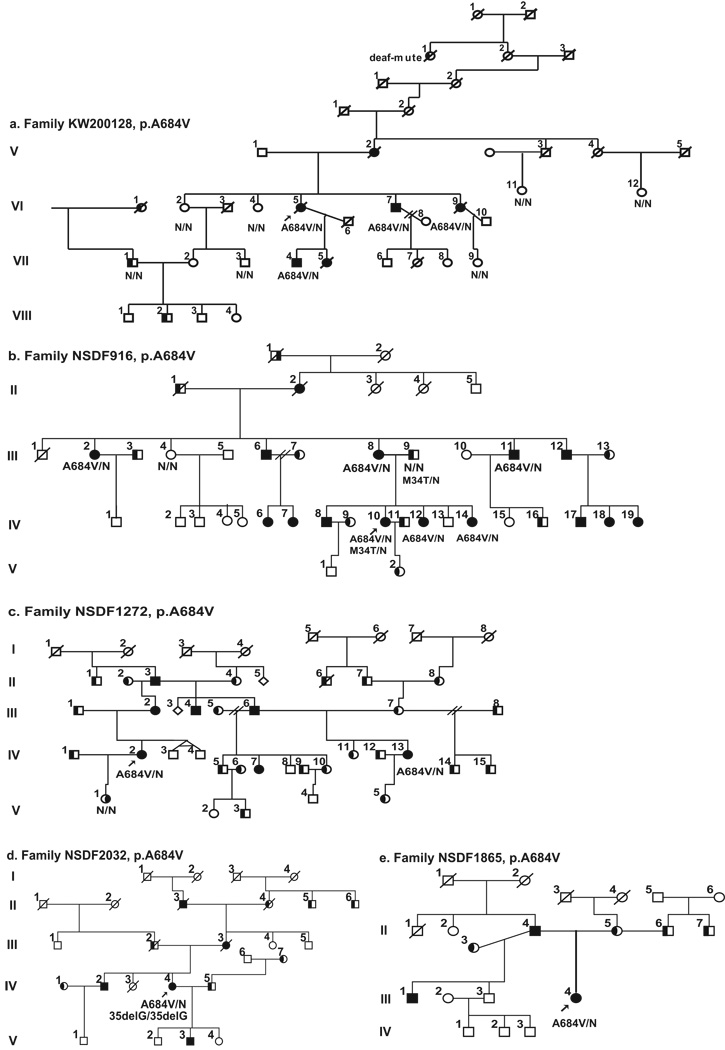
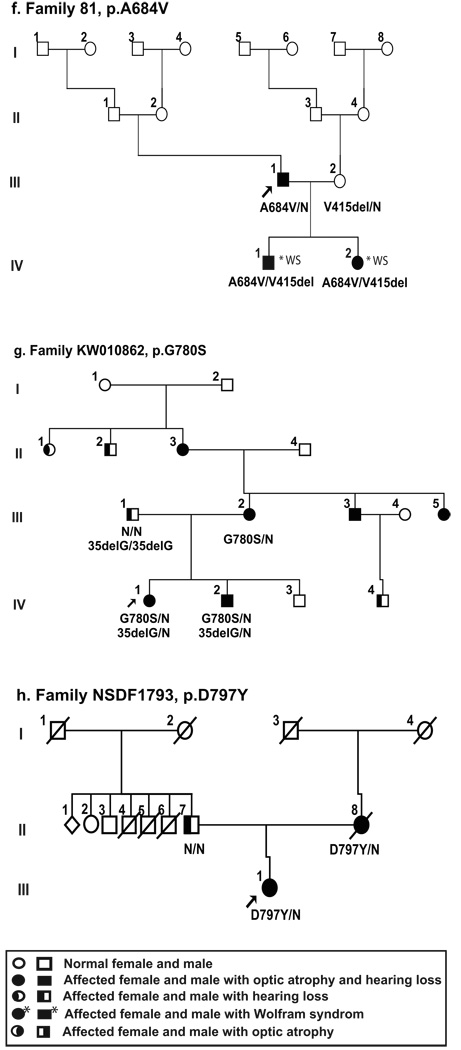
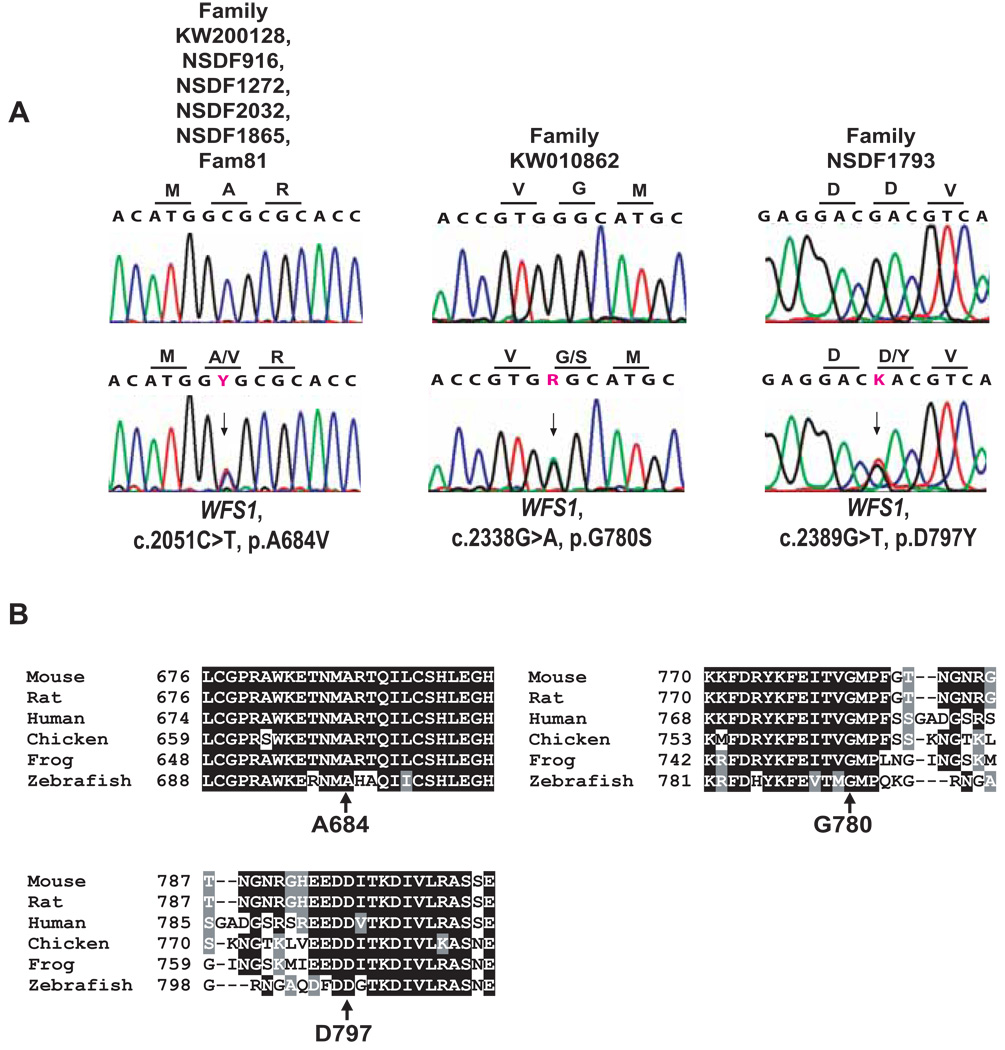
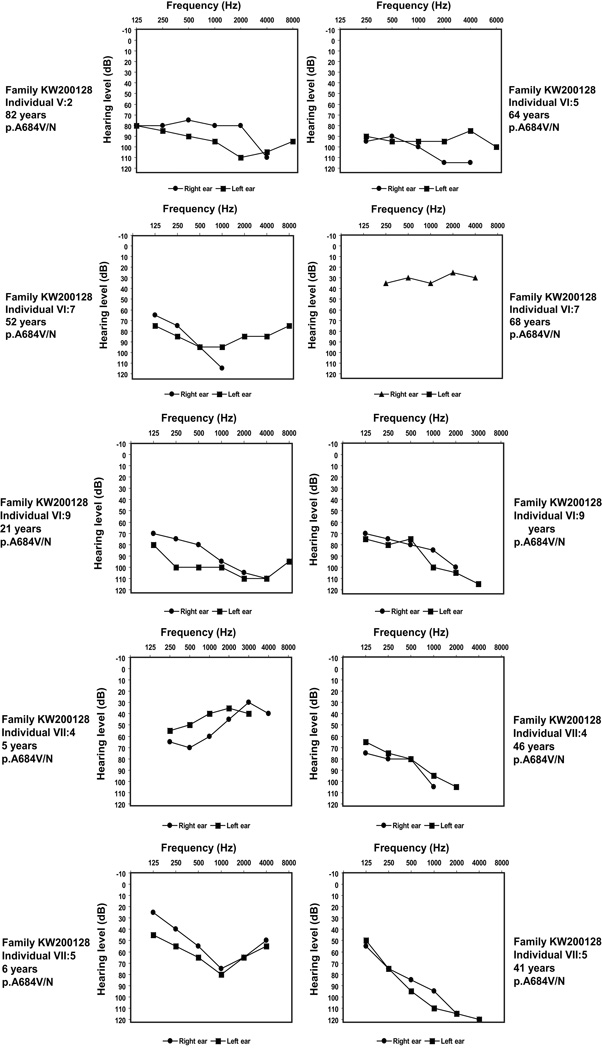
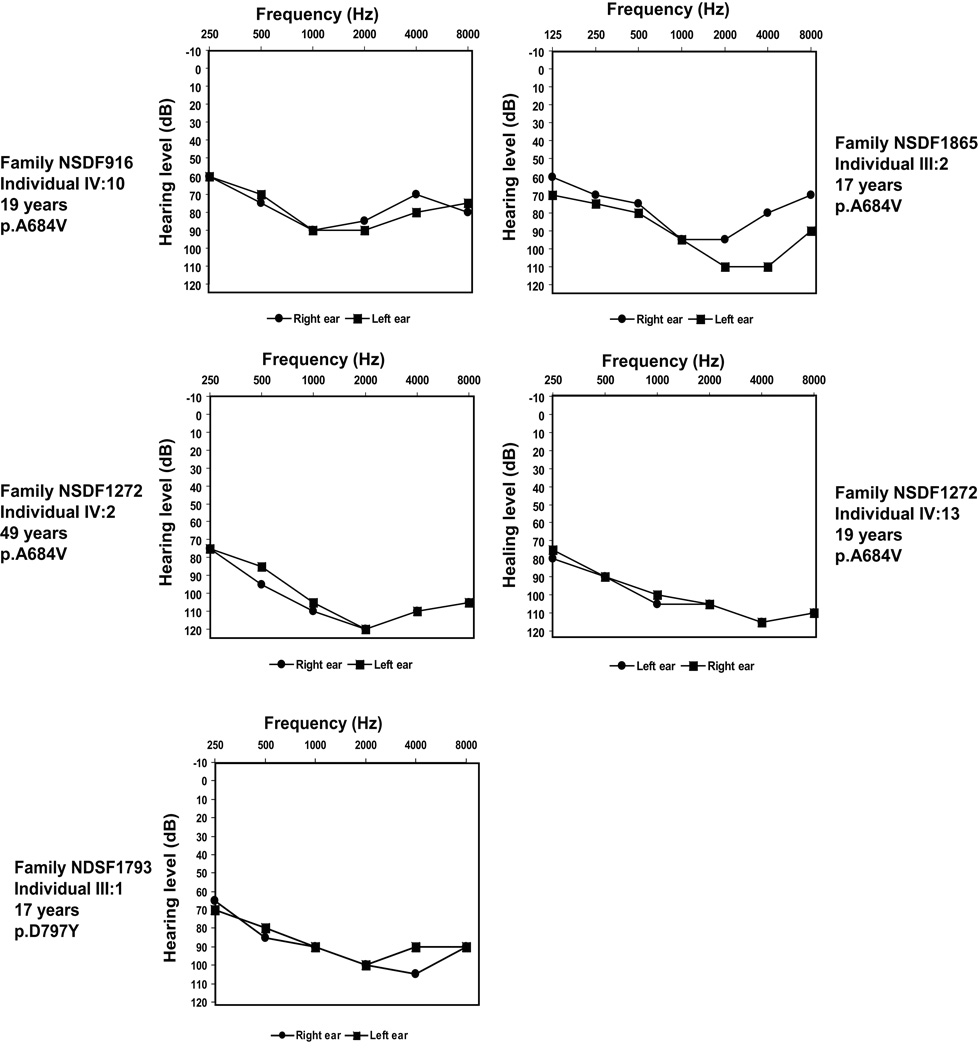
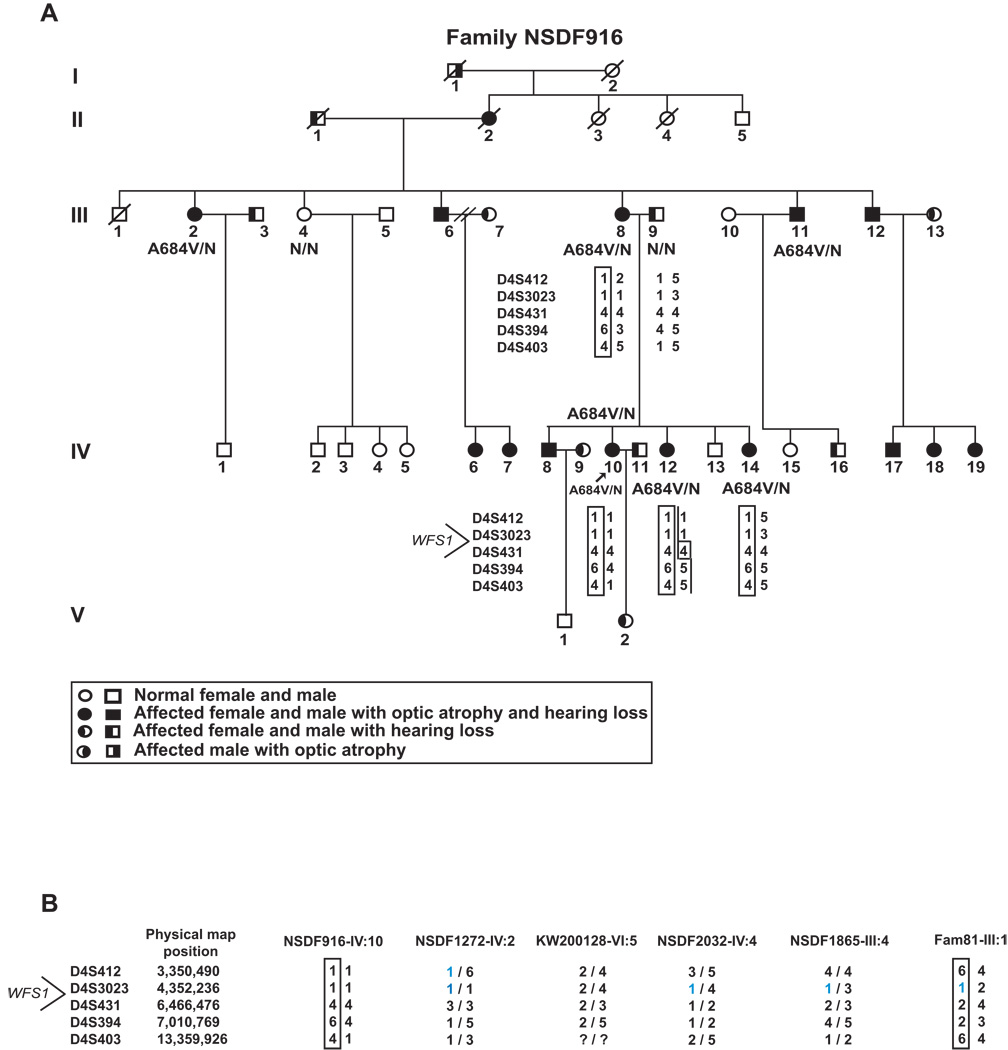
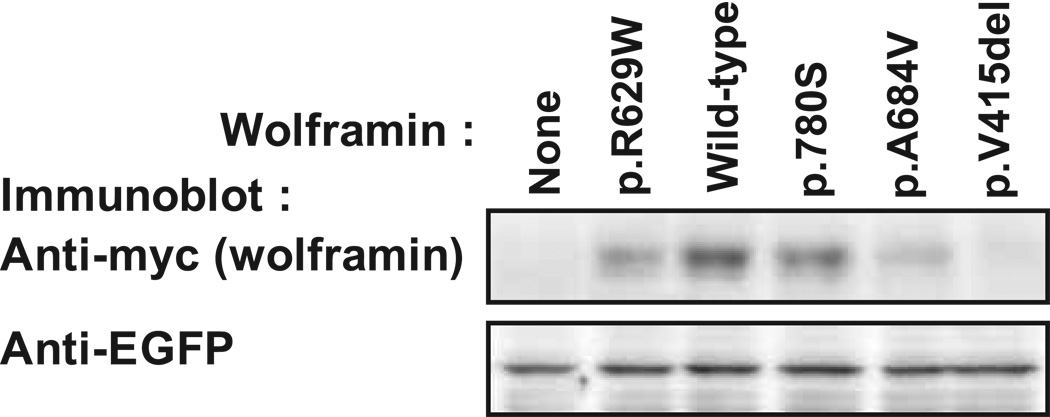
Optic atrophy (OA) and sensorineural hearing loss (SNHL) are key abnormalities in several syndromes, including the recessively inherited Wolfram syndrome, caused by mutations in WFS1. In contrast, the association of autosomal dominant OA and SNHL without other phenotypic abnormalities is rare, and almost exclusively attributed to mutations in the Optic Atrophy-1 gene (OPA1), most commonly the p.R445H mutation. We present eight probands and their families from the US, Sweden, and UK with OA and SNHL, whom we analyzed for mutations in OPA1 and WFS1. Among these families, we found three heterozygous missense mutations in WFS1 segregating with OA and SNHL: p.A684V (six families), and two novel mutations, p.G780S and p.D797Y, all involving evolutionarily conserved amino acids and absent from 298 control chromosomes. Importantly, none of these families harbored the OPA1 p.R445H mutation. No mitochondrial DNA deletions were detected in muscle from one p.A684V patient analyzed. Finally, wolframin p.A684V mutant ectopically expressed in HEK cells showed reduced protein levels compared to wild-type wolframin, strongly indicating that the mutation is disease-causing. Our data support OA and SNHL as a phenotype caused by dominant mutations in WFS1 in these additional eight families. Importantly, our data provide the first evidence that a single, recurrent mutation in WFS1, p.A684V, may be a common cause of ADOA and SNHL, similar to the role played by the p.R445H mutation in OPA1. Our findings suggest that patients who are heterozygous for WFS1 missense mutations should be carefully clinically examined for OA and other manifestations of Wolfram syndrome.
Copyright © 2011 Wiley-Liss, Inc.
Figures
References
-
- Amati-Bonneau P, Guichet A, Olichon A, Chevrollier A, Viala F, Miot S, Ayuso C, Odent S, Arrouet C, Verny C, Calmels MN, Simard G, Belenguer P, Wang J, Puel JL, Hamel C, Malthiery Y, Bonneau D, Lenaers G, Reynier P. OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Ann Neurol. 2005;58:958–963. - PubMed
-
- Amati-Bonneau P, Milea D, Bonneau D, Chevrollier A, Ferre M, Guillet V, Gueguen N, Loiseau D, de Crescenzo MA, Verny C, Procaccio V, Lenaers G, Reynier P. OPA1-associated disorders: phenotypes and pathophysiology. Int J Biochem Cell Biol. 2009;41:1855–1865. - PubMed
-
- Amati-Bonneau P, Odent S, Derrien C, Pasquier L, Malthiery Y, Reynier P, Bonneau D. The association of autosomal dominant optic atrophy and moderate deafness may be due to the R445H mutation in the OPA1 gene. Am J Ophthalmol. 2003;136:1170–1171. - PubMed
-
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, Labauge P, Figarella-Branger D, Marcorelles P, Furby A, Beauvais K, Letournel F, Liguori R, La MC, Montagna P, Liguori M, Zanna C, Rugolo M, Cossarizza A, Wissinger B, Verny C, Schwarzenbacher R, Martin MA, Arenas J, Ayuso C, Garesse R, Lenaers G, Bonneau D, Carelli V. OPA1 mutations induce mitochondrial DNA instability and optic atrophy 'plus' phenotypes. Brain. 2008;131:338–351. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
