

Hydrogen-bonding catalysis and inhibition by simple solvents in the stereoselective kinetic epoxide-opening spirocyclization of glycal epoxides to form spiroketals
- PMID: 21539313
- PMCID: PMC3113711
- DOI: 10.1021/ja201249c
Hydrogen-bonding catalysis and inhibition by simple solvents in the stereoselective kinetic epoxide-opening spirocyclization of glycal epoxides to form spiroketals
Abstract
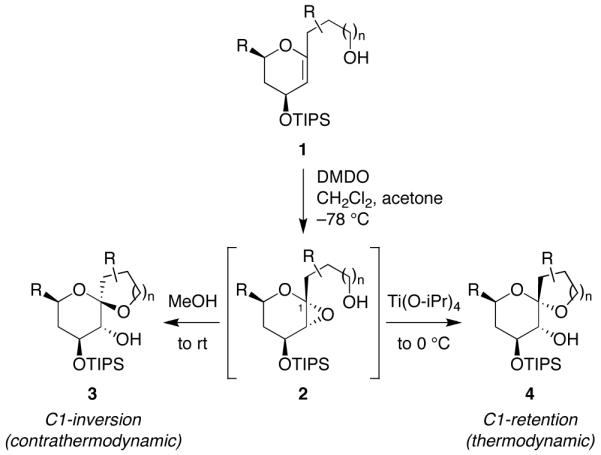
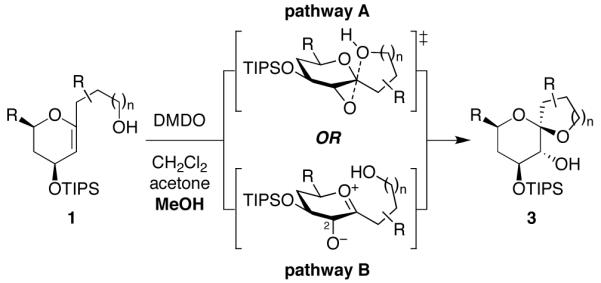
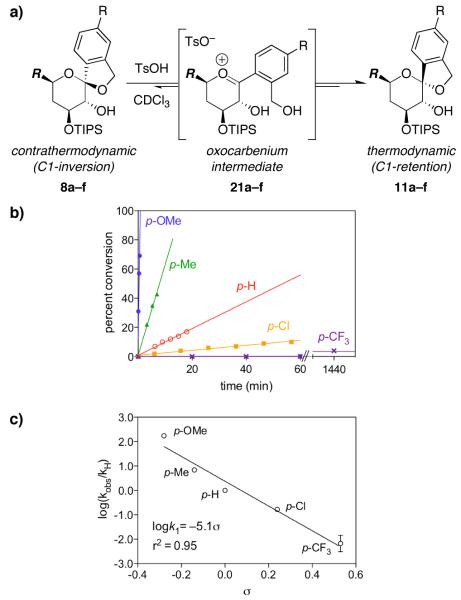
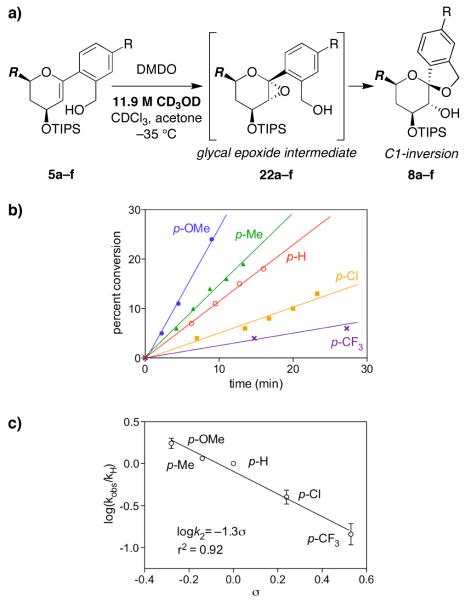
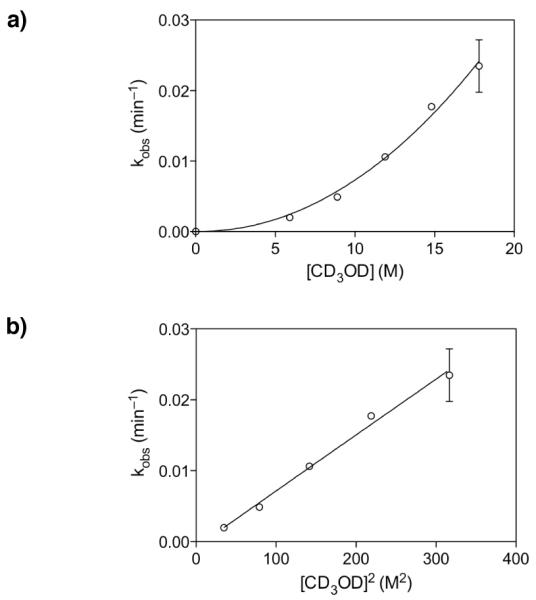
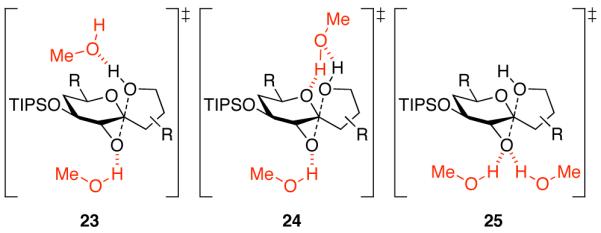
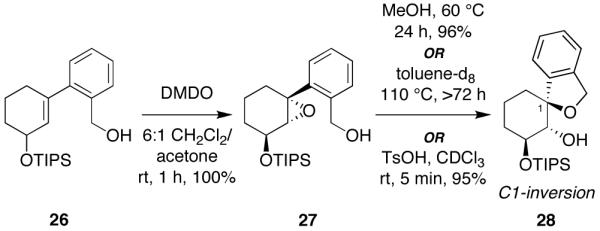
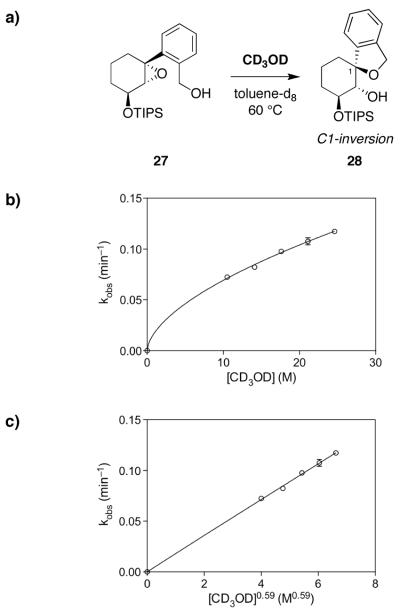
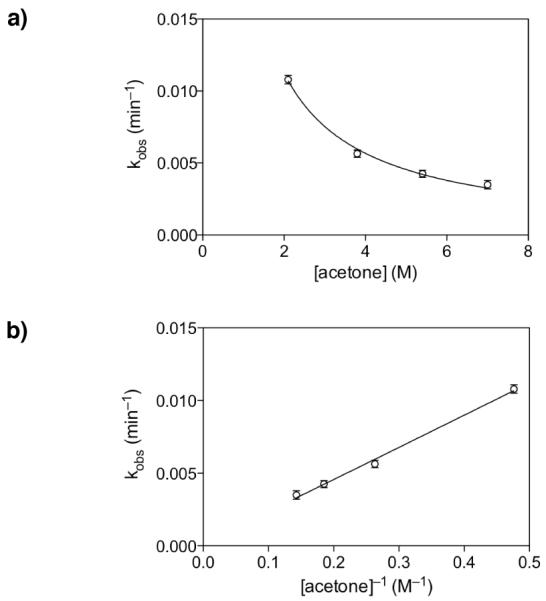
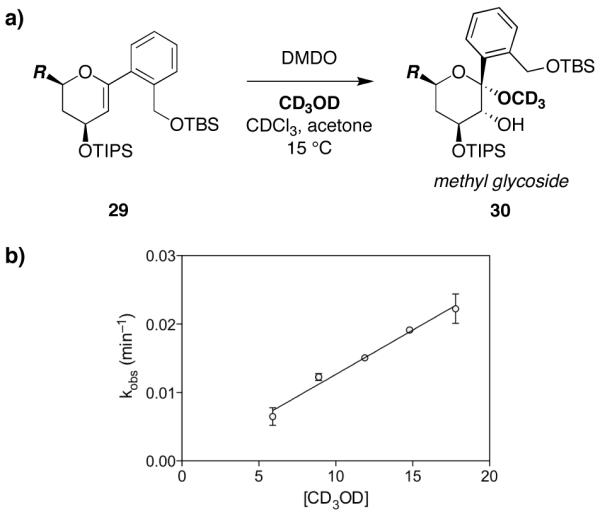
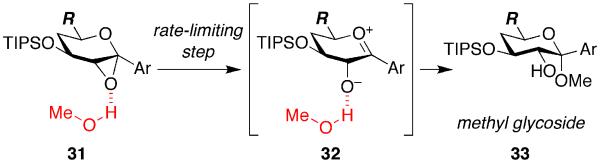
Mechanistic investigations of a MeOH-induced kinetic epoxide-opening spirocyclization of glycal epoxides have revealed dramatic, specific roles for simple solvents in hydrogen-bonding catalysis of this reaction to form spiroketal products stereoselectively with inversion of configuration at the anomeric carbon. A series of electronically tuned C1-aryl glycal epoxides was used to study the mechanism of this reaction based on differential reaction rates and inherent preferences for S(N)2 versus S(N)1 reaction manifolds. Hammett analysis of reaction kinetics with these substrates is consistent with an S(N)2 or S(N)2-like mechanism (ρ = -1.3 vs ρ = -5.1 for corresponding S(N)1 reactions of these substrates). Notably, the spirocyclization reaction is second-order dependent on MeOH, and the glycal ring oxygen is required for second-order MeOH catalysis. However, acetone cosolvent is a first-order inhibitor of the reaction. A transition state consistent with the experimental data is proposed in which one equivalent of MeOH activates the epoxide electrophile via a hydrogen bond while a second equivalent of MeOH chelates the side-chain nucleophile and glycal ring oxygen. A paradoxical previous observation that decreased MeOH concentration leads to increased competing intermolecular methyl glycoside formation is resolved by the finding that this side reaction is only first-order dependent on MeOH. This study highlights the unusual abilities of simple solvents to act as hydrogen-bonding catalysts and inhibitors in epoxide-opening reactions, providing both stereoselectivity and discrimination between competing reaction manifolds. This spirocyclization reaction provides efficient, stereocontrolled access to spiroketals that are key structural motifs in natural products.
Figures
References
-
- Wassermann A. J. Chem. Soc. 1942:618–621.
- Yates P, Eaton P. J. Am. Chem. Soc. 1960;82:4436–4437.
-
- Hine J, Linden S-M, Kanagasabapathy VM. J. Org. Chem. 1985;50:5096–5099.
-
- Potuzak JS, Moilanen SB, Tan DS. J. Am. Chem. Soc. 2005;127:13796–13797. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
