

A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis
- PMID: 21540242
- PMCID: PMC3180233
- DOI: 10.1242/dmm.007237
A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis
Abstract
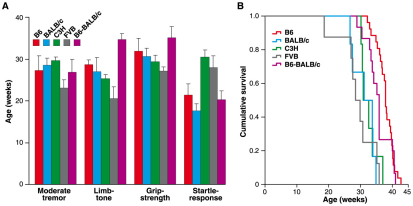
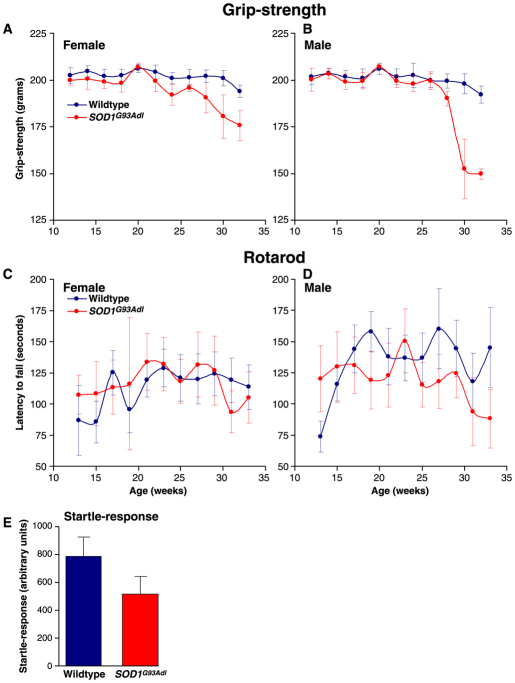
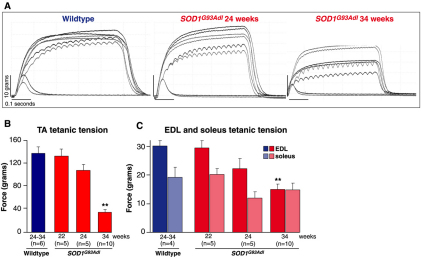
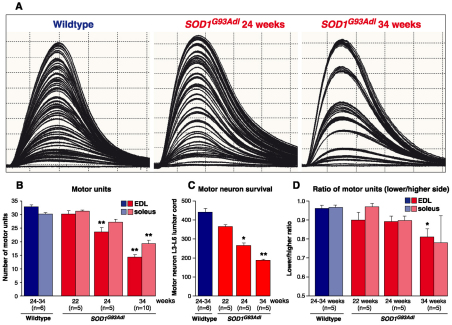
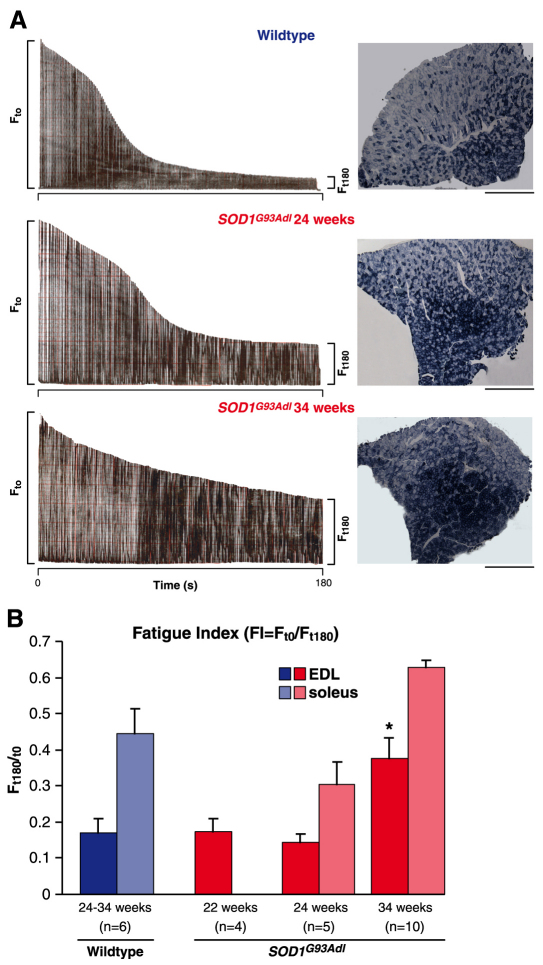
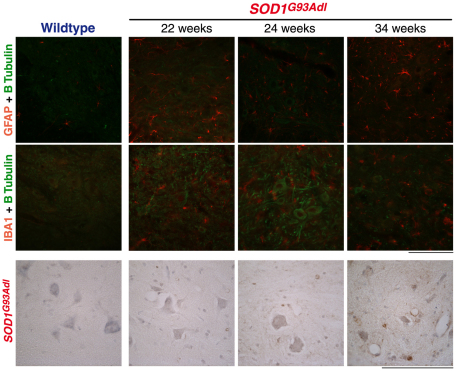
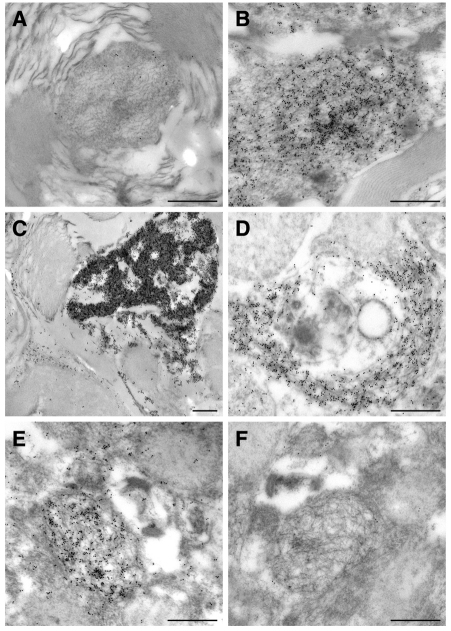
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that results in the death of motor neurons in the brain and spinal cord. The disorder generally strikes in mid-life, relentlessly leading to paralysis and death, typically 3-5 years after diagnosis. No effective treatments are available. Up to 10% of ALS is familial, usually autosomal dominant. Several causative genes are known and, of these, mutant superoxide dismutase 1 (SOD1) is by far the most frequently found, accounting for up to 20% of familial ALS. A range of human mutant SOD1 transgenic mouse strains has been produced, and these largely successfully model the human disease. Of these, the most widely used is the SOD1 mouse, which expresses a human SOD1 transgene with a causative G93A mutation. This mouse model is excellent for many purposes but carries up to 25 copies of the transgene and produces a great excess of SOD1 protein, which might affect our interpretation of disease processes. A variant of this strain carries a deletion of the transgene array such that the copy number is dropped to eight to ten mutant SOD1 genes. This 'deleted' 'low-copy' mouse undergoes a slower course of disease, over many months. Here we have carried out a comprehensive analysis of phenotype, including nerve and muscle physiology and histology, to add to our knowledge of this 'deleted' strain and give baseline data for future studies. We find differences in phenotype that arise from genetic background and sex, and we quantify the loss of nerve and muscle function over time. The slowly progressive pathology observed in this mouse strain could provide us with a more appropriate model for studying early-stage pathological processes in ALS and aid the development of therapies for early-stage treatments.
Figures
References
-
- Achilli F., Boyle S., Kieran D., Chia R., Hafezparast M., Martin J. E., Schiavo G., Greensmith L., Bickmore W., Fisher E. M. (2005). The SOD1 transgene in the G93A mouse model of amyotrophic lateral sclerosis lies on distal mouse chromosome 12. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 6, 111–114 - PubMed
-
- Alexander G. M., Erwin K. L., Byers N., Deitch J. S., Augelli B. J., Blankenhorn E. P., Heiman-Patterson T. D. (2004). Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res. Mol. Brain Res. 130, 7–15 - PubMed
-
- Avraham K. B., Schickler M., Sapoznikov D., Yarom R., Groner Y. (1988). Down’s syndrome: abnormal neuromuscular junction in tongue of transgenic mice with elevated levels of human Cu/Zn-superoxide dismutase. Cell 54, 823–829 - PubMed
-
- Benatar M. (2007). Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol. Dis. 26, 1–13 - PubMed
-
- Birve A., Neuwirth C., Weber M., Marklund S. L., Nilsson A. C., Jonsson P. A., Andersen P. M. (2010). A novel SOD1 splice site mutation associated with familial ALS revealed by SOD activity analysis. Hum. Mol. Genet. 19, 4201–4206 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
