

Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism
- PMID: 21541297
- PMCID: PMC3082572
- DOI: 10.1371/journal.pone.0019315
Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism
Abstract
Restriction-site associated DNA (RAD) sequencing is a powerful new method for targeted sequencing across the genomes of many individuals. This approach has broad potential for genetic analysis of non-model organisms including genotype-phenotype association mapping, phylogeography, population genetics and scaffolding genome assemblies through linkage mapping. We constructed a RAD library using genomic DNA from a Plutella xylostella (diamondback moth) backcross that segregated for resistance to the insecticide spinosad. Sequencing of 24 individuals was performed on a single Illumina GAIIx lane (51 base paired-end reads). Taking advantage of the lack of crossing over in homologous chromosomes in female Lepidoptera, 3,177 maternally inherited RAD alleles were assigned to the 31 chromosomes, enabling identification of the spinosad resistance and W/Z sex chromosomes. Paired-end reads for each RAD allele were assembled into contigs and compared to the genome of Bombyx mori (n = 28) using BLAST, revealing 28 homologous matches plus 3 expected fusion/breakage events which account for the difference in chromosome number. A genome-wide linkage map (1292 cM) was inferred with 2,878 segregating RAD alleles inherited from the backcross father, producing chromosome and location specific sequenced RAD markers. Here we have used RAD sequencing to construct a genetic linkage map de novo for an organism that has no previous genome data. Comparative analysis of P. xyloxtella linkage groups with B. mori chromosomes shows for the first time, genetic synteny appears common beyond the Macrolepidoptera. RAD sequencing is a powerful system capable of rapidly generating chromosome specific data for non-model organisms.
Conflict of interest statement
Figures
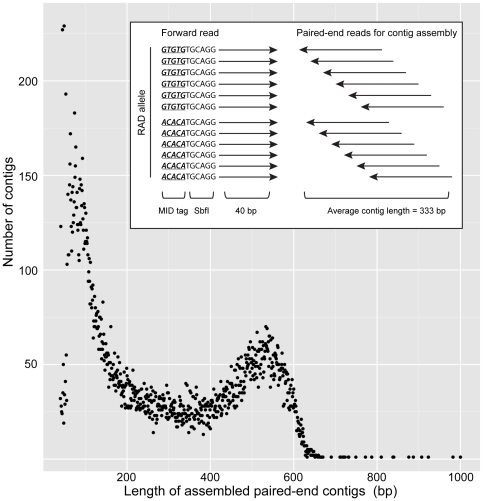
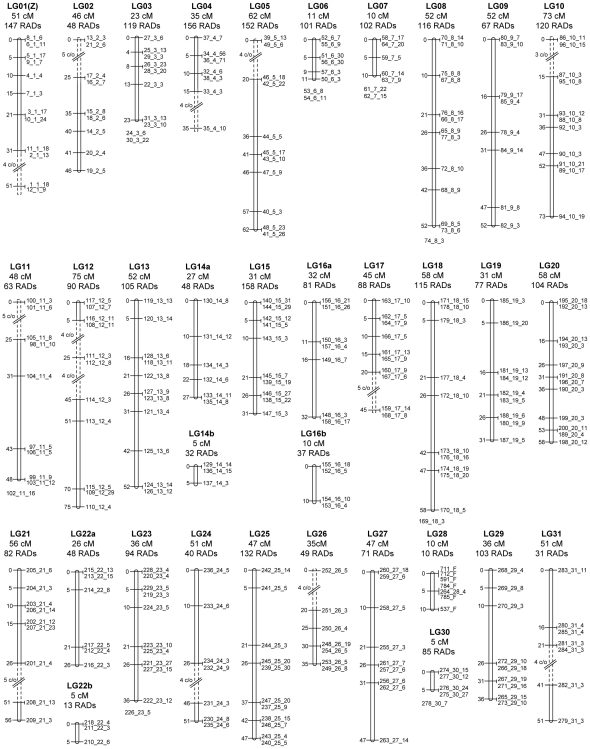
References
-
- Colosimo P, Hosemann K, Balabhadra S, Villarreal GJ, Dickson M, et al. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science. 2005;307:1928–1933. - PubMed
-
- Peichel CL, Nereng KS, Ohgi KA, Cole BLE, Colosimo PF, et al. The genetic architecture of divergence between threespine stickleback species. Nature. 2001;414:901–905. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- G0900740/MRC_/Medical Research Council/United Kingdom
- BB/E021107/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- RG47616/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- F021135/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- G00661X/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
