

Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome
- PMID: 21541312
- PMCID: PMC3082526
- DOI: 10.1371/journal.pone.0018441
Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome
Abstract
Background: Neisseria meningitidis is a naturally transformable, facultative pathogen colonizing the human nasopharynx. Here, we analyze on a genome-wide level the impact of recombination on gene-complement diversity and virulence evolution in N. meningitidis. We combined comparative genome hybridization using microarrays (mCGH) and multilocus sequence typing (MLST) of 29 meningococcal isolates with computational comparison of a subset of seven meningococcal genome sequences.
Principal findings: We found that lateral gene transfer of minimal mobile elements as well as prophages are major forces shaping meningococcal population structure. Extensive gene content comparison revealed novel associations of virulence with genetic elements besides the recently discovered meningococcal disease associated (MDA) island. In particular, we identified an association of virulence with a recently described canonical genomic island termed IHT-E and a differential distribution of genes encoding RTX toxin- and two-partner secretion systems among hyperinvasive and non-hyperinvasive lineages. By computationally screening also the core genome for signs of recombination, we provided evidence that about 40% of the meningococcal core genes are affected by recombination primarily within metabolic genes as well as genes involved in DNA replication and repair. By comparison with the results of previous mCGH studies, our data indicated that genetic structuring as revealed by mCGH is stable over time and highly similar for isolates from different geographic origins.
Conclusions: Recombination comprising lateral transfer of entire genes as well as homologous intragenic recombination has a profound impact on meningococcal population structure and genome composition. Our data support the hypothesis that meningococcal virulence is polygenic in nature and that differences in metabolism might contribute to virulence.
Conflict of interest statement
Figures
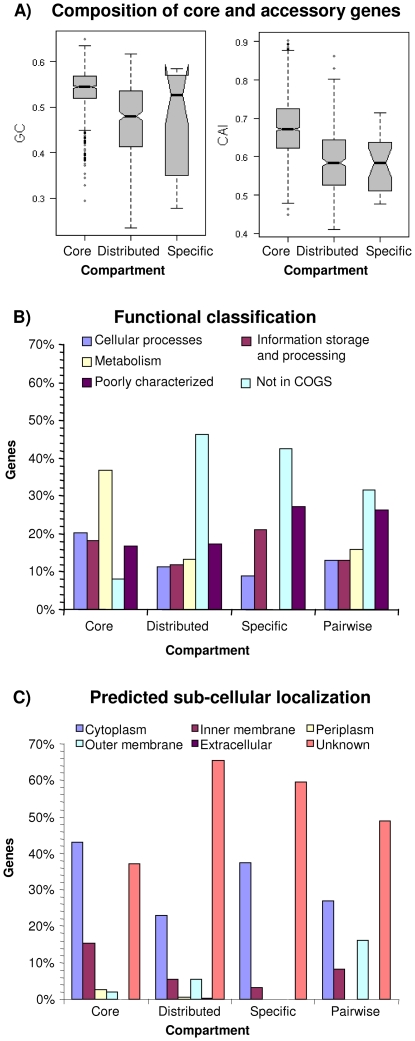
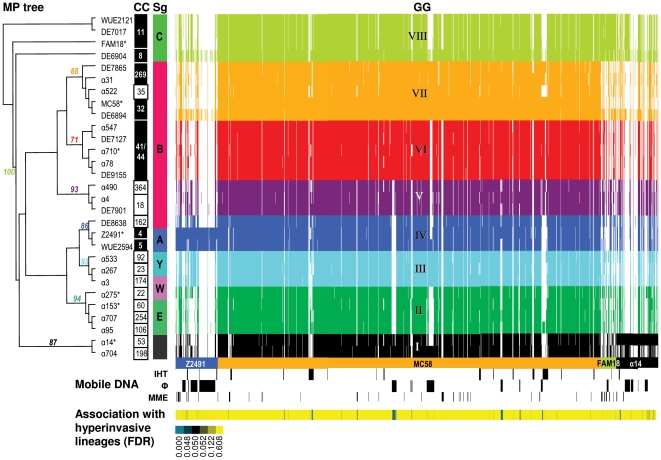
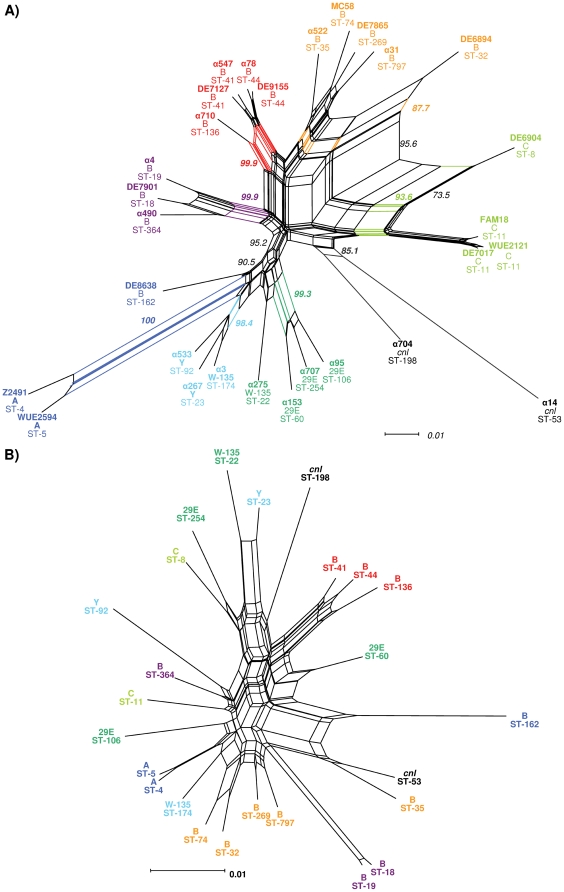
References
-
- Claus H, Maiden MC, Wilson DJ, McCarthy ND, Jolley KA, et al. Genetic analysis of meningococci carried by children and young adults. J Infect Dis. 2005;191:1263–1271. - PubMed
-
- Stephens DS, Greenwood B, Brandtzaeg P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet. 2007;369:2196–2210. - PubMed
-
- Frosch M, Vogel U. Structure and genetics of the meningococcal capsule. In: Frosch M, Maiden MC, editors. Handbook of Meningococcal Disease. Weinheim, Germany: Wiley-VCH; 2006. pp. 145–162.
-
- Harrison LH, Trotter CL, Ramsay ME. Global epidemiology of meningococcal disease. Vaccine. 2009;27(Suppl 2):B51–63. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
