

Clinical manifestations and treatment of mucopolysaccharidosis type I patients in Latin America as compared with the rest of the world
- PMID: 21541721
- PMCID: PMC3173625
- DOI: 10.1007/s10545-011-9336-2
Clinical manifestations and treatment of mucopolysaccharidosis type I patients in Latin America as compared with the rest of the world
Abstract
Background: Mucopolysaccharidosis I (MPS I) comprises a spectrum of clinical manifestations and is divided into three phenotypes reflecting clinical severity: Hurler, Hurler-Scheie, and Scheie syndromes. There may be important variations in clinical manifestations of this genetic disease in patients residing in different regions of the world.
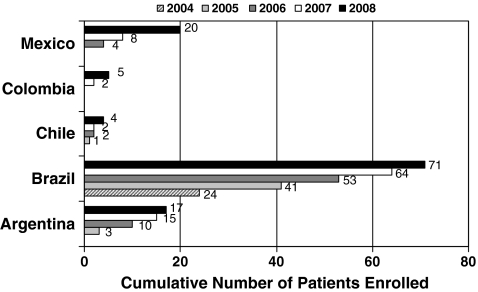
Methods: Using data from the MPS I Registry (as of September 2009), we evaluated patients from Latin America (n = 118) compared with patients from the rest of the world [ROW (n = 727)].
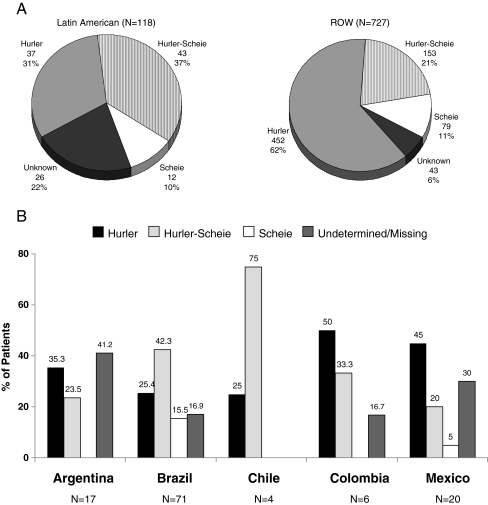
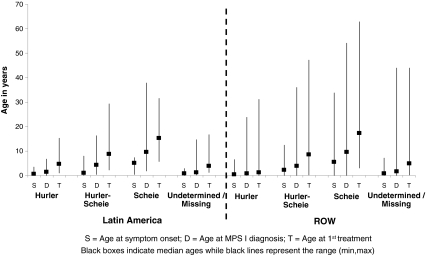
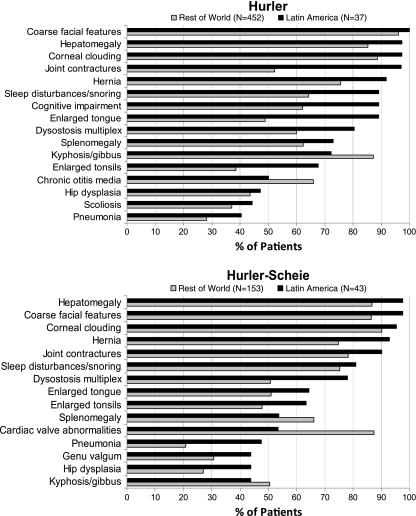
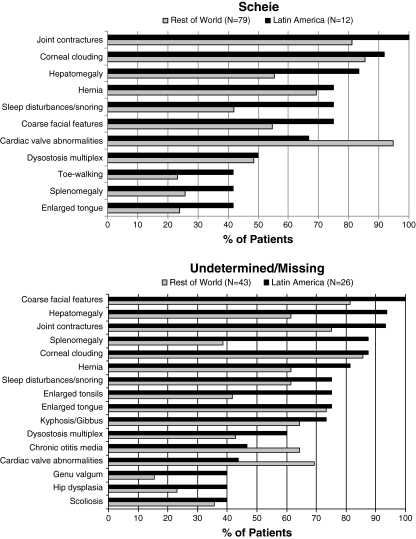
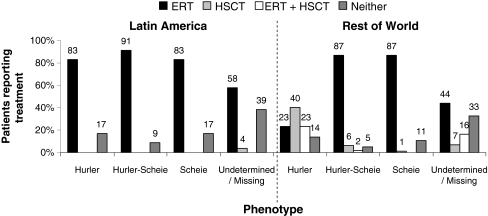
Results: Phenotype distribution differed among patients in Latin America compared to ROW (Hurler 31 vs. 62%, Hurler-Scheie 36 vs. 21%, Scheie 10 vs. 11%, and unknown 22 vs. 6%). The frequency of certain symptoms, such as cardiac valve abnormalities, sleep impairment, and joint contractures, also differed between Latin America and ROW for some phenotypes. Median age at MPS I diagnosis was earlier in the ROW than Latin America for all phenotypes, and age at first treatment for Hurler and Hurler-Scheie patients was also earlier in the ROW. Hurler patients in Latin America showed a gap of 3.1 years between median ages of diagnosis and first treatment compared to only 0.5 years in the ROW. Treatment allocation in Latin America compared to ROW was as follows: enzyme replacement therapy (ERT) only, 80 vs. 45%; hematopoietic stem cell transplantation (HSCT) only, 0.9 vs. 27%; both ERT and HSCT, 0 vs. 16%; and neither treatment, 19 vs. 13%.
Conclusion: These data highlight important differences in MPS I patients between Latin America and ROW in terms of phenotypic distribution, clinical manifestations, and treatment practices.
Conflict of interest statement
The authors declare independence from the sponsors and declare that the content of the article has not been influenced by the sponsors.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
