

Activation of Akt rescues endoplasmic reticulum stress-impaired murine cardiac contractile function via glycogen synthase kinase-3β-mediated suppression of mitochondrial permeation pore opening
- PMID: 21542787
- PMCID: PMC3176349
- DOI: 10.1089/ars.2010.3751
Activation of Akt rescues endoplasmic reticulum stress-impaired murine cardiac contractile function via glycogen synthase kinase-3β-mediated suppression of mitochondrial permeation pore opening
Retraction in
-
Retraction of: Activation of Akt Rescues Endoplasmic Reticulum Stress-Impaired Murine Cardiac Contractile Function via Glycogen Synthase Kinase-3β-Mediated Suppression of Mitochondrial Permeation Pore Opening (Antioxid Redox Signal 15(9);2011:2407-2424; doi: 10.1089/ars.2010.3751).Antioxid Redox Signal. 2023 Jun;38(16-18):1214. doi: 10.1089/ars.2010.3751.retract. Epub 2023 Apr 18. Antioxid Redox Signal. 2023. PMID: 37074108 Free PMC article. No abstract available.
Abstract
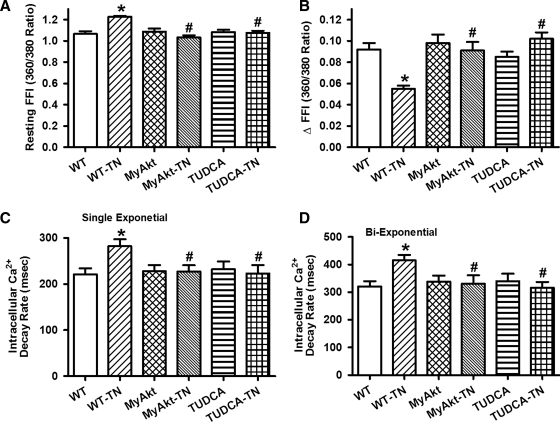
Aims: The present study was designed to examine the impact of chronic Akt activation on endoplasmic reticulum (ER) stress-induced cardiac mechanical anomalies, if any, and the underlying mechanism involved.
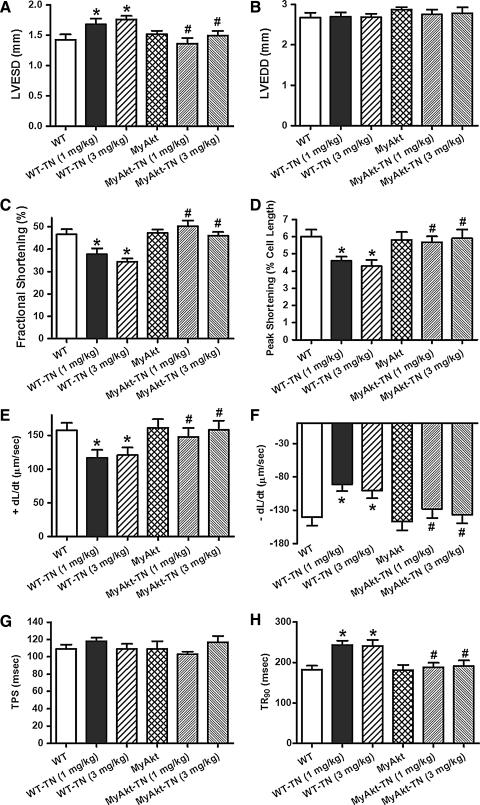
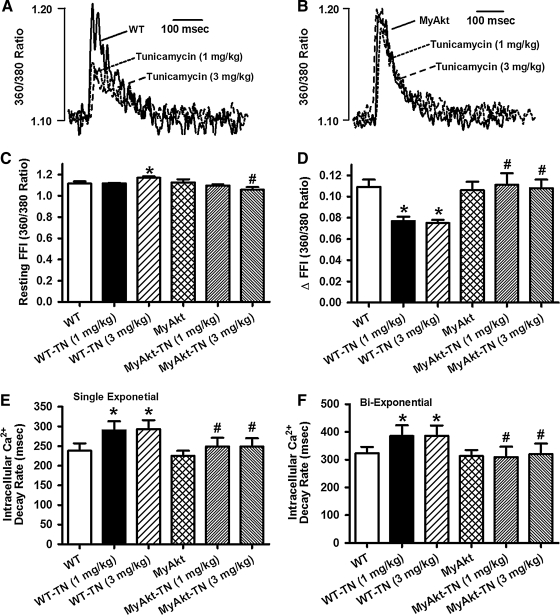
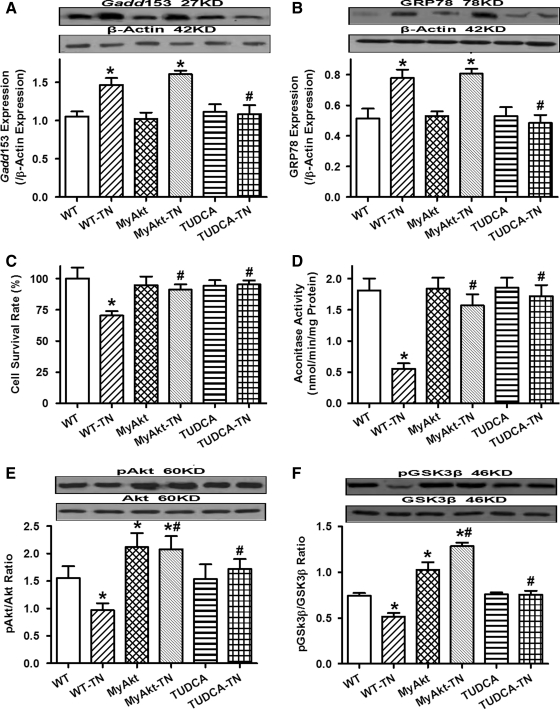
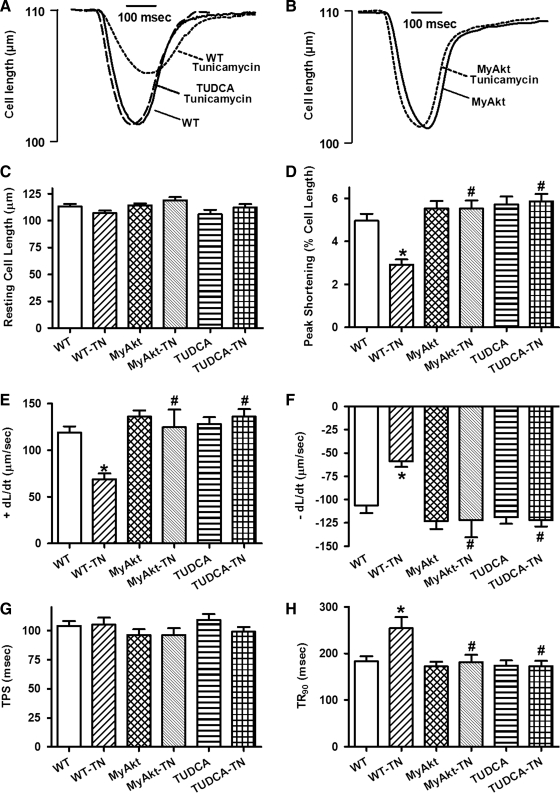
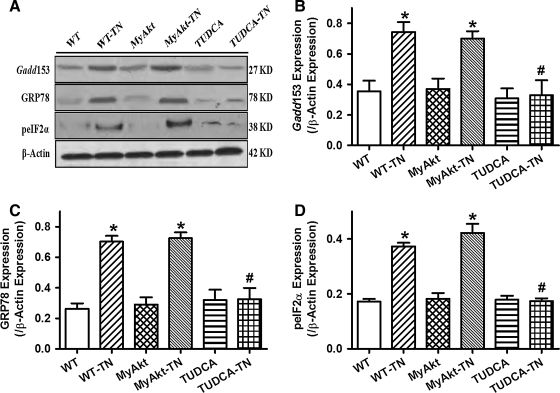
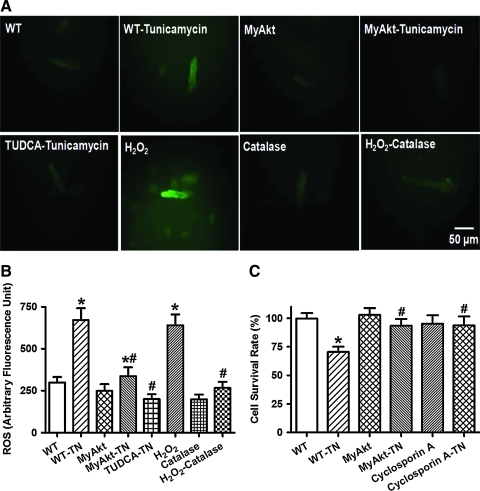
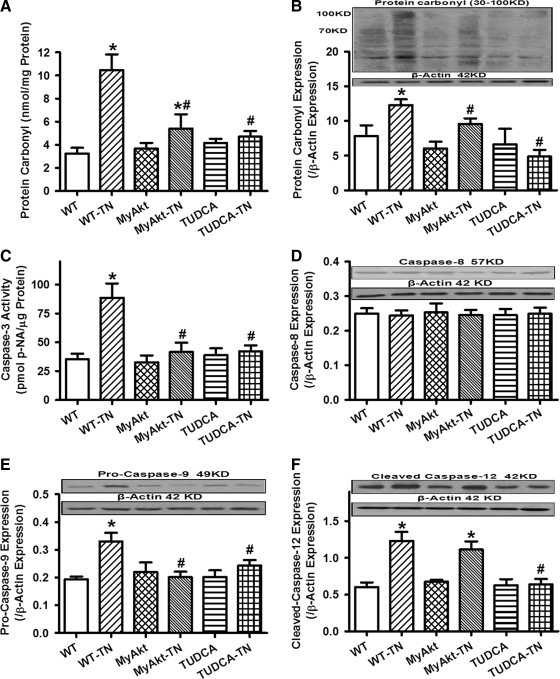
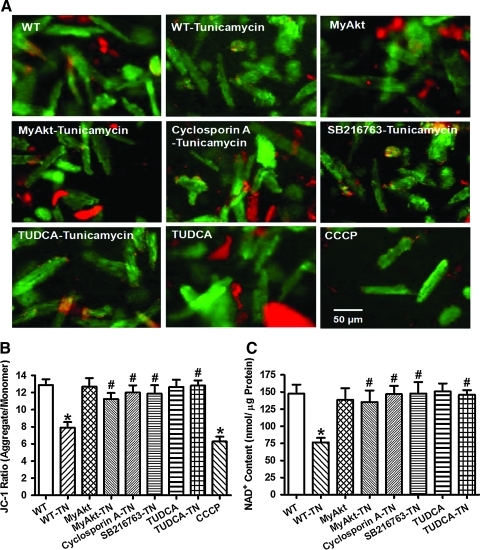
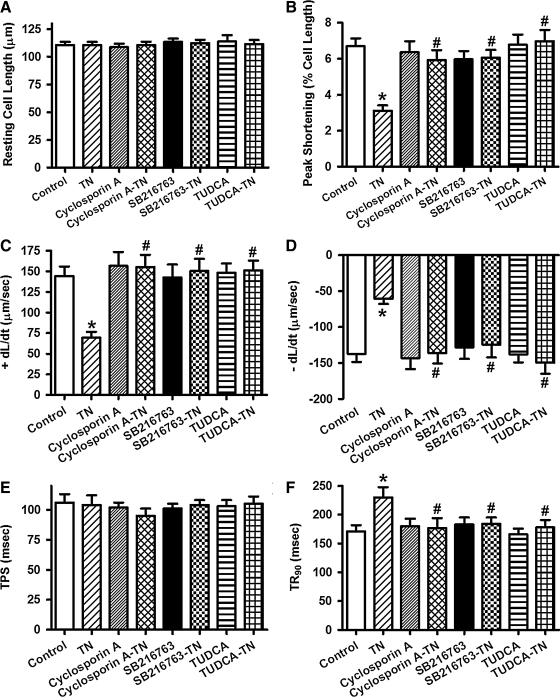
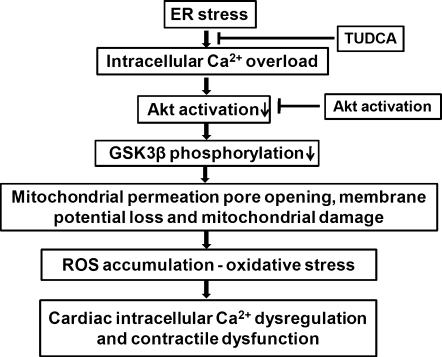
Results: Wild-type and transgenic mice with cardiac-specific overexpression of the active mutant of Akt (Myr-Akt) were subjected to the ER stress inducer tunicamycin (1 or 3 mg/kg). ER stress led to compromised echocardiographic (elevated left ventricular end-systolic diameter and reduced fractional shortening) and cardiomyocyte contractile function, intracellular Ca(2+) mishandling, and cell survival in wild-type mice associated with mitochondrial damage. In vitro ER stress induction in murine cardiomyocytes upregulated the ER stress proteins Gadd153, GRP78, and phospho-eIF2α, and promoted reactive oxygen species production, carbonyl formation, apoptosis, mitochondrial membrane potential loss, and mitochondrial permeation pore (mPTP) opening associated with overtly impaired cardiomyocyte contractile and intracellular Ca(2+) properties. Interestingly, these anomalies were mitigated by chronic Akt activation or the ER chaperon tauroursodeoxycholic acid (TUDCA). Treatment with tunicamycin also dephosphorylated Akt and its downstream signal glycogen synthase kinase 3β (GSK3β) (leading to activation of GSK3β), the effect of which was abrogated by Akt activation and TUDCA. The ER stress-induced cardiomyocyte contractile and mitochondrial anomalies were obliterated by the mPTP inhibitor cyclosporin A, GSK3β inhibitor SB216763, and ER stress inhibitor TUDCA.
Innovation: This research reported the direct relationship between ER stress and cardiomyocyte contractile and mitochondrial anomalies for the first time.
Conclusion: Taken together, these data suggest that ER stress may compromise cardiac contractile and intracellular Ca(2+) properties, possibly through the Akt/GSK3β-dependent impairment of mitochondrial integrity.
Figures
Similar articles
-
Chronic Akt activation attenuated lipopolysaccharide-induced cardiac dysfunction via Akt/GSK3β-dependent inhibition of apoptosis and ER stress.Biochim Biophys Acta. 2013 Jun;1832(6):848-63. doi: 10.1016/j.bbadis.2013.02.023. Epub 2013 Mar 6. Biochim Biophys Acta. 2013. PMID: 23474308 Free PMC article.
-
RETRACTED: Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: Role of Akt dephosphorylation.Free Radic Biol Med. 2011 Dec 15;51(12):2172-2184. doi: 10.1016/j.freeradbiomed.2011.09.005. Epub 2011 Sep 16. Free Radic Biol Med. 2011. Retraction in: Free Radic Biol Med. 2023 Nov 1;208:877. doi: 10.1016/j.freeradbiomed.2023.09.025. PMID: 21996563 Free PMC article. Retracted.
-
Tauroursodeoxycholic acid mitigates high fat diet-induced cardiomyocyte contractile and intracellular Ca2+ anomalies.PLoS One. 2013 May 7;8(5):e63615. doi: 10.1371/journal.pone.0063615. Print 2013. PLoS One. 2013. PMID: 23667647 Free PMC article.
-
Mitochondrial Oxidative Stress Is the General Reason for Apoptosis Induced by Different-Valence Heavy Metals in Cells and Mitochondria.Int J Mol Sci. 2023 Sep 22;24(19):14459. doi: 10.3390/ijms241914459. Int J Mol Sci. 2023. PMID: 37833908 Free PMC article. Review.
-
Mitochondria play a central role in nonischemic cardiomyocyte necrosis: common to acute and chronic stressor states.Pflugers Arch. 2012 Jul;464(1):123-31. doi: 10.1007/s00424-012-1079-x. Epub 2012 Feb 11. Pflugers Arch. 2012. PMID: 22328074 Free PMC article. Review.
Cited by
-
Does Transient Receptor Potential Vanilloid Type 1 Alleviate or Aggravate Pathological Myocardial Hypertrophy?Front Pharmacol. 2021 May 10;12:681286. doi: 10.3389/fphar.2021.681286. eCollection 2021. Front Pharmacol. 2021. PMID: 34040539 Free PMC article. Review.
-
Heavy metal scavenger metallothionein attenuates ER stress-induced myocardial contractile anomalies: role of autophagy.Toxicol Lett. 2014 Mar 21;225(3):333-41. doi: 10.1016/j.toxlet.2013.12.024. Epub 2014 Jan 17. Toxicol Lett. 2014. PMID: 24440343 Free PMC article.
-
The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases.Glob Adv Health Med. 2014 May;3(3):58-69. doi: 10.7453/gahmj.2014.017. Glob Adv Health Med. 2014. PMID: 24891994 Free PMC article. Review.
-
Retraction Note: Mitochondrial aldehyde dehydrogenase (ALDH2) protects against streptozotocin-induced diabetic cardiomyopathy: role of GSK3β and mitochondrial function.BMC Med. 2022 Jul 8;20(1):248. doi: 10.1186/s12916-022-02455-5. BMC Med. 2022. PMID: 35804375 Free PMC article. No abstract available.
-
Mitochondrial Ca2+ regulation in the etiology of heart failure: physiological and pathophysiological implications.Acta Pharmacol Sin. 2020 Oct;41(10):1301-1309. doi: 10.1038/s41401-020-0476-5. Epub 2020 Jul 21. Acta Pharmacol Sin. 2020. PMID: 32694759 Free PMC article. Review.
References
-
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. - PubMed
-
- Chien KR. Stress pathways and heart failure. Cell. 1999;98:555–558. - PubMed
-
- Deniaud A. Sharaf El DO. Maillier E. Poncet D. Kroemer G. Lemaire C. Brenner C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27:285–299. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
