

Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq
- PMID: 21543516
- PMCID: PMC3129258
- DOI: 10.1101/gr.110882.110
Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq
Abstract
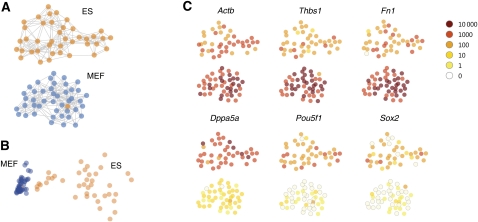
Our understanding of the development and maintenance of tissues has been greatly aided by large-scale gene expression analysis. However, tissues are invariably complex, and expression analysis of a tissue confounds the true expression patterns of its constituent cell types. Here we describe a novel strategy to access such complex samples. Single-cell RNA-seq expression profiles were generated, and clustered to form a two-dimensional cell map onto which expression data were projected. The resulting cell map integrates three levels of organization: the whole population of cells, the functionally distinct subpopulations it contains, and the single cells themselves-all without need for known markers to classify cell types. The feasibility of the strategy was demonstrated by analyzing the transcriptomes of 85 single cells of two distinct types. We believe this strategy will enable the unbiased discovery and analysis of naturally occurring cell types during development, adult physiology, and disease.
Figures
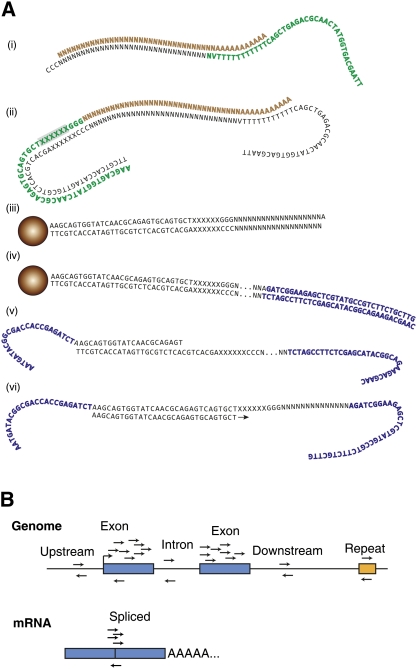
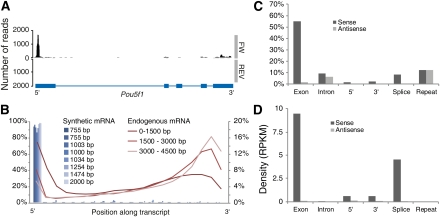
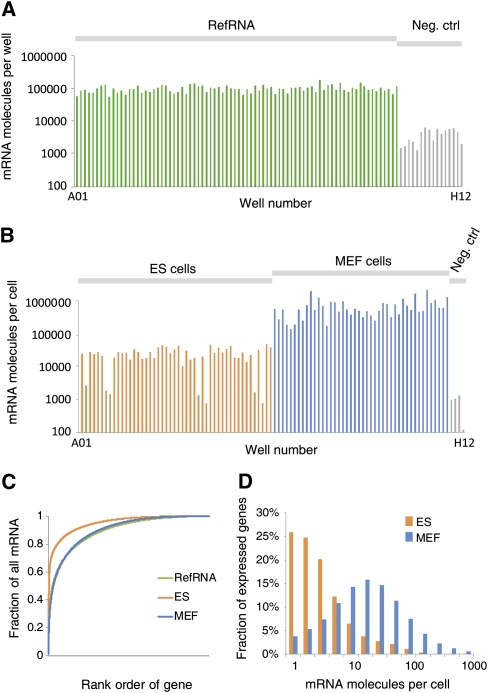
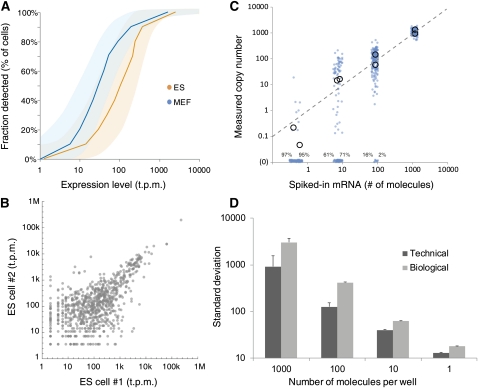
References
-
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G et al. 2008. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5: 613–619 - PubMed
-
- Eisenberg E, Levanon EY 2003. Human housekeeping genes are compact. Trends Genet 19: 362–365 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases