

Extensive epigenetic reprogramming in human somatic tissues between fetus and adult
- PMID: 21545704
- PMCID: PMC3112062
- DOI: 10.1186/1756-8935-4-7
Extensive epigenetic reprogramming in human somatic tissues between fetus and adult
Abstract
Background: Development of human tissue is influenced by a combination of intrinsic biological signals and extrinsic environmental stimuli, both of which are mediated by epigenetic regulation, including DNA methylation. However, little is currently known of the normal acquisition or loss of epigenetic markers during fetal and postnatal development.
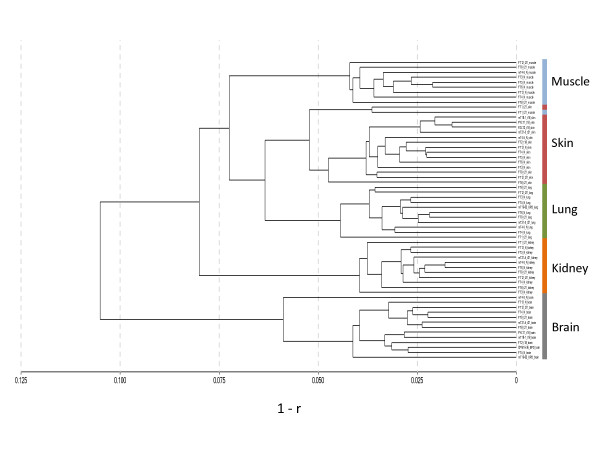
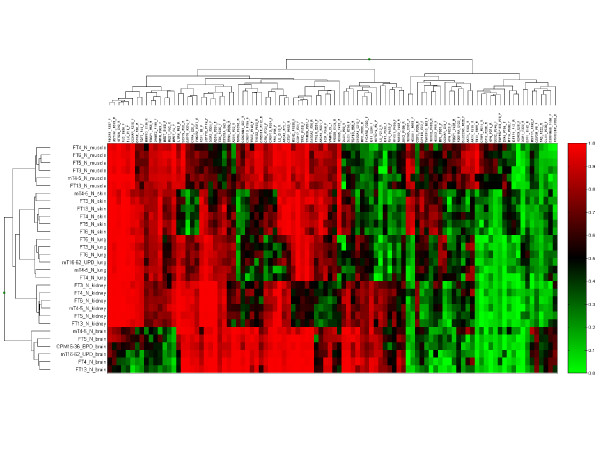
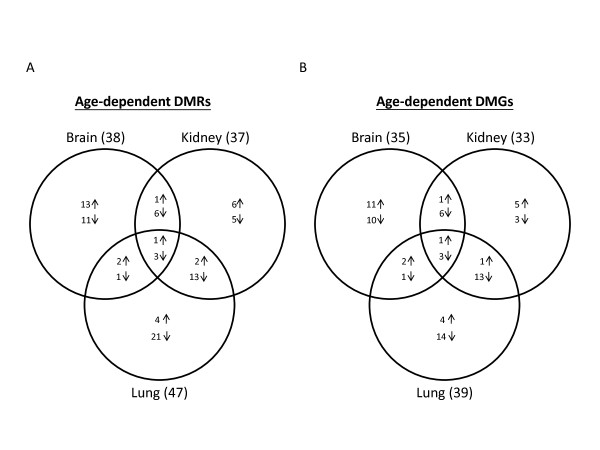
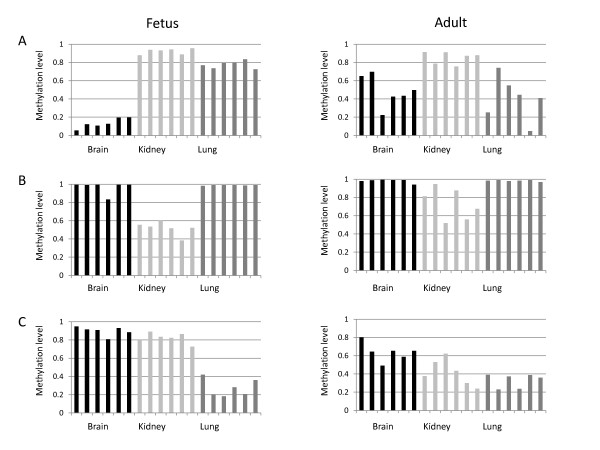
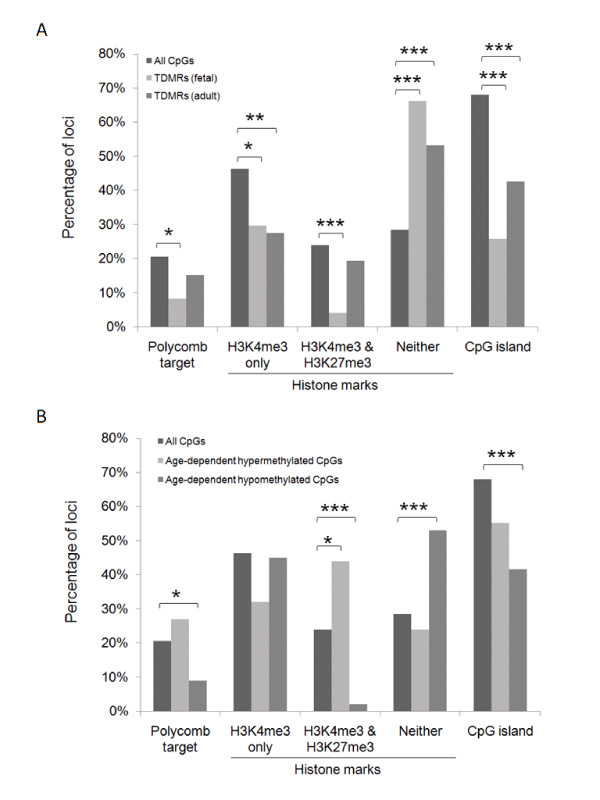
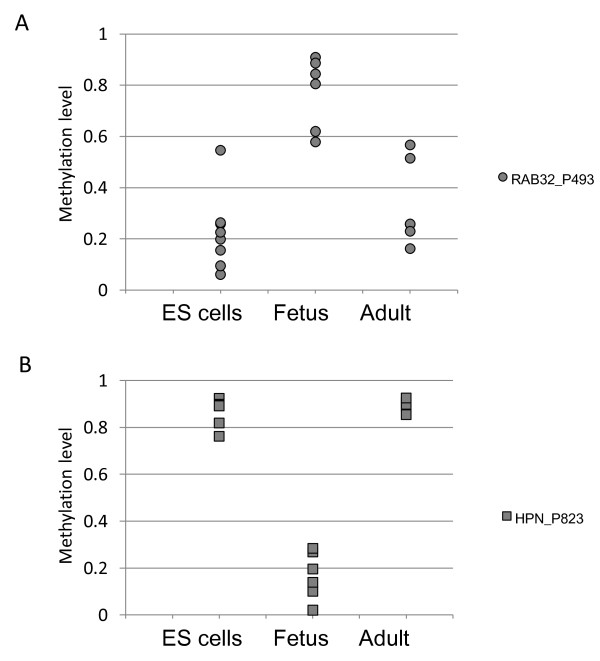
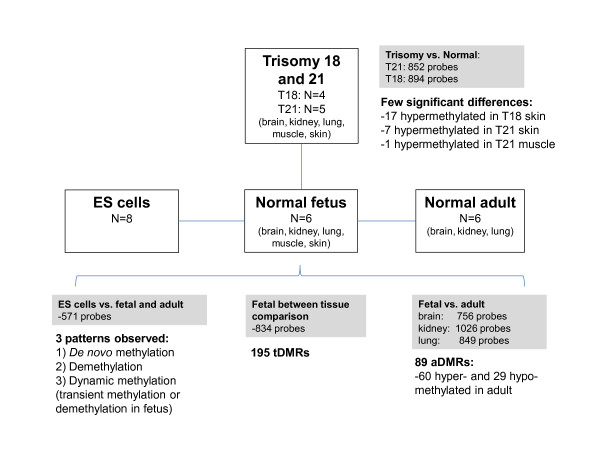
Results: The DNA methylation status of over 1000 CpGs located in the regulatory regions of nearly 800 genes was evaluated in five somatic tissues (brain, kidney, lung, muscle and skin) from eight normal second-trimester fetuses. Tissue-specific differentially methylated regions (tDMRs) were identified in 195 such loci. However, comparison with corresponding data from trisomic fetuses (five trisomy 21 and four trisomy 18) revealed relatively few DNA methylation differences associated with trisomy, despite such conditions having a profound effect on development. Of interest, only 17% of the identified fetal tDMRs were found to maintain this same tissue-specific DNA methylation in adult tissues. Furthermore, 10% of the sites analyzed, including sites associated with imprinted genes, had a DNA methylation difference of >40% between fetus and adult. This plasticity of DNA methylation over development was further confirmed by comparison with similar data from embryonic stem cells, with the most altered methylation levels being linked to domains with bivalent histone modifications.
Conclusions: Most fetal tDMRs seem to reflect transient DNA methylation changes during development rather than permanent epigenetic signatures. The extensive tissue-specific and developmental-stage specific nature of DNA methylation will need to be elucidated to identify abnormal patterns of DNA methylation associated with abnormal development or disease.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
