

Af17 deficiency increases sodium excretion and decreases blood pressure
- PMID: 21546577
- PMCID: PMC3103727
- DOI: 10.1681/ASN.2010121270
Af17 deficiency increases sodium excretion and decreases blood pressure
Abstract
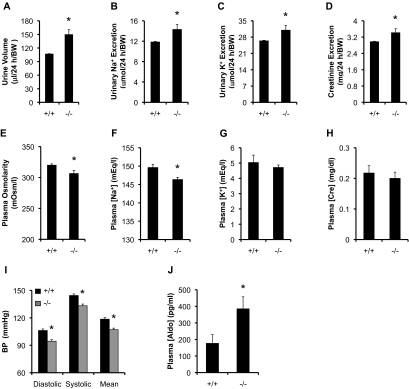
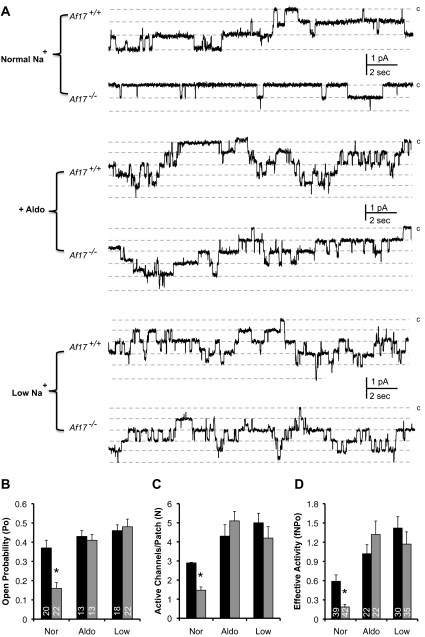
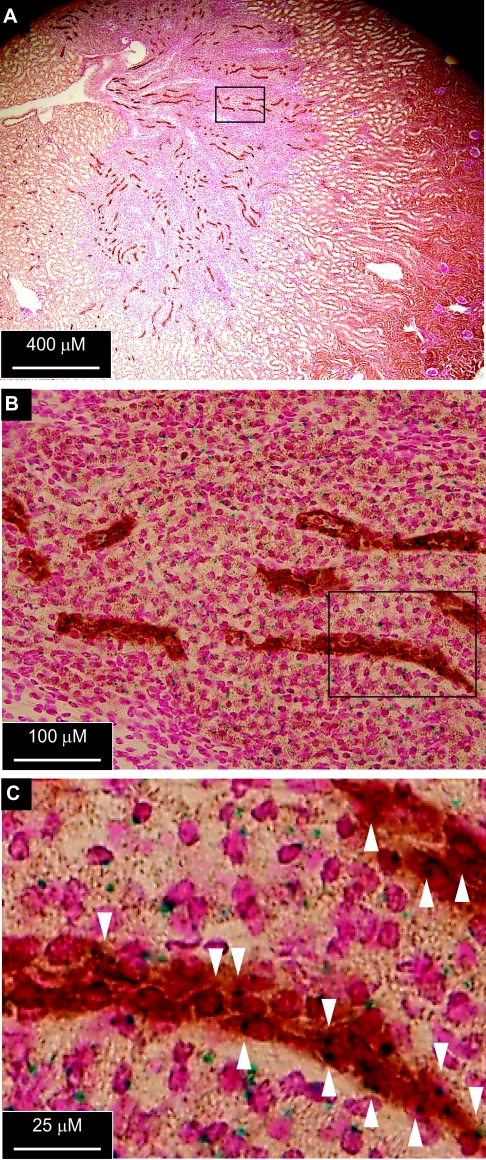
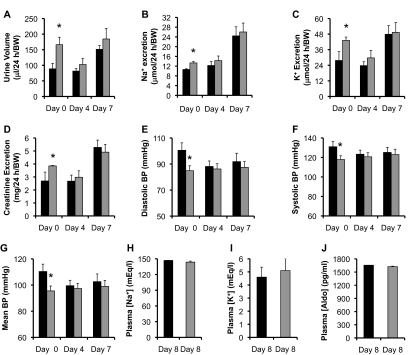
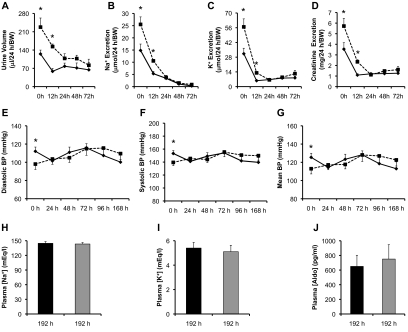
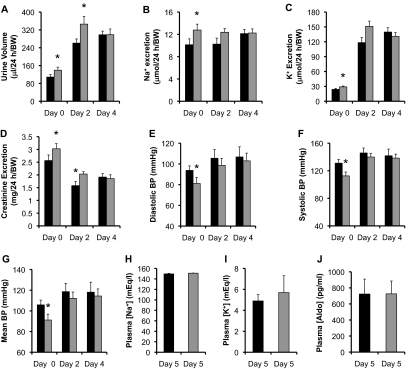
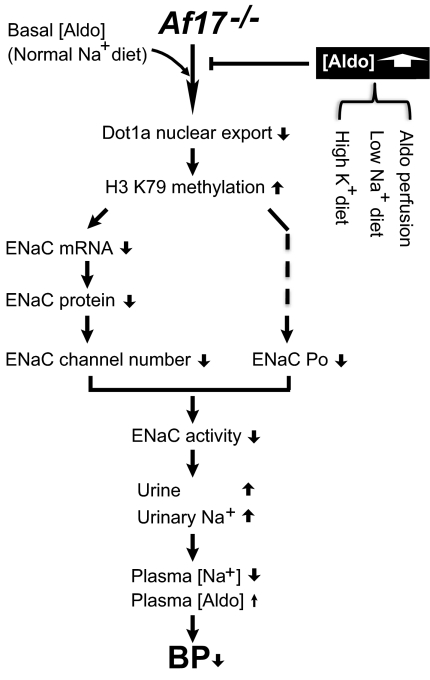
The putative transcription factor AF17 upregulates the transcription of the epithelial sodium channel (ENaC) genes, but whether AF17 modulates sodium homeostasis and BP is unknown. Here, we generated Af17-deficient mice to determine whether deletion of Af17 leads to sodium wasting and low BP. Compared with wild-type mice, Af17-deficient mice had lower BP (11 mmHg), higher urine volume, and increased sodium excretion despite mildly increased plasma concentrations of aldosterone. Deletion of Af17 led to increased dimethylation of histone H3 K79 and reduced ENaC function. The attenuated function of ENaC resulted from decreased ENaC mRNA and protein expression, fewer active channels, lower open probability, and reduced effective activity. In contrast, inducing high levels of plasma aldosterone by a variety of methods completely compensated for Af17 deficiency with respect to sodium handling and BP. Taken together, these data identify Af17 as a potential locus for the maintenance of sodium and BP homeostasis and suggest that a particular histone modification is directly linked to these processes. Af17-mediated regulation of BP is largely, but not exclusively, the result of modulating ENaC, suggesting it has potential as a therapeutic target for the control of BP.
Figures


References
-
- Wilson RC, Krozowski ZS, Li K, Obeyesekere VR, Razzaghy-Azar M, Harbison MD, Wei JQ, Shackleton CH, Funder JW, New MI: A mutation in the HSD11B2 gene in a family with apparent mineralocorticoid excess. J Clin Endocrinol Metab 80: 2263–2266, 1995 - PubMed
-
- Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, Lalouel JM: A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 355: 262–265, 1992 - PubMed
-
- Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, Meinke G, Tsai FT, Sigler PB, Lifton RP: Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science 289: 119–123, 2000 - PubMed
-
- Rafestin-Oblin ME, Souque A, Bocchi B, Pinon G, Fagart J, Vandewalle A: The severe form of hypertension caused by the activating S810L mutation in the mineralocorticoid receptor is cortisone related. Endocrinology 144: 528–533, 2003 - PubMed
-
- Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP: Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
