

cAMP-guanine exchange factor protection from bile acid-induced hepatocyte apoptosis involves glycogen synthase kinase regulation of c-Jun NH2-terminal kinase
- PMID: 21546580
- PMCID: PMC3280825
- DOI: 10.1152/ajpgi.00430.2010
cAMP-guanine exchange factor protection from bile acid-induced hepatocyte apoptosis involves glycogen synthase kinase regulation of c-Jun NH2-terminal kinase
Abstract
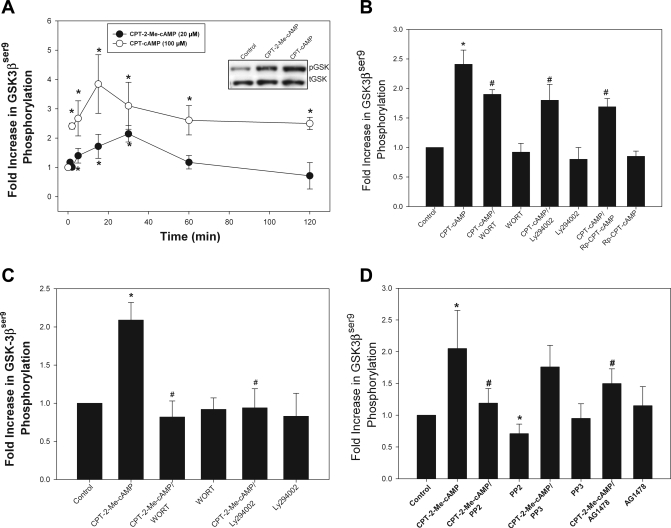
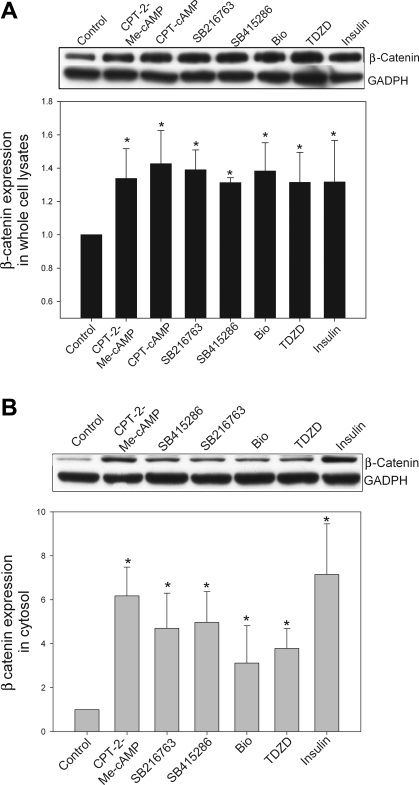
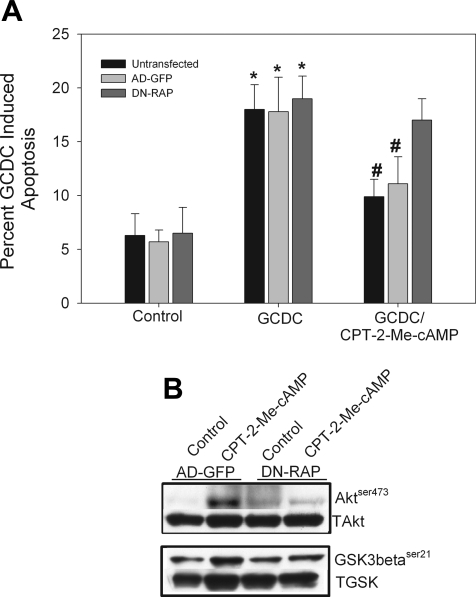
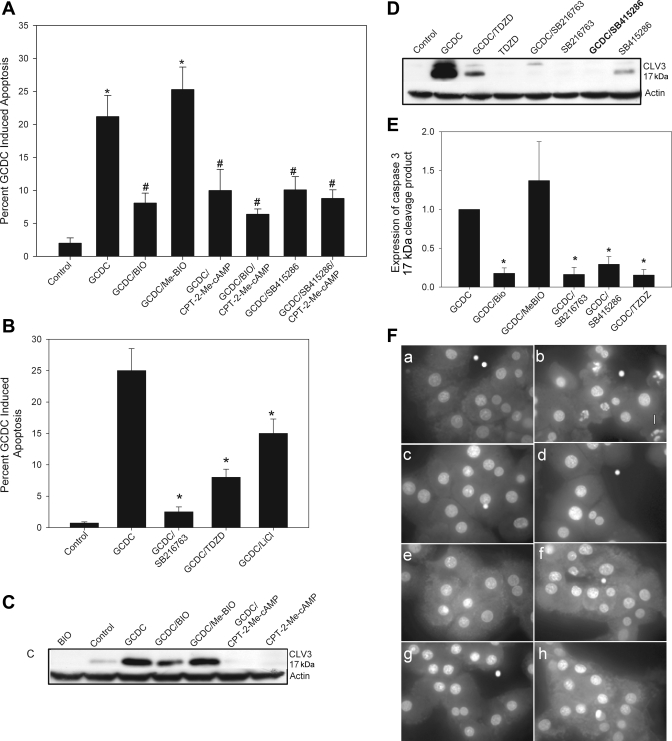
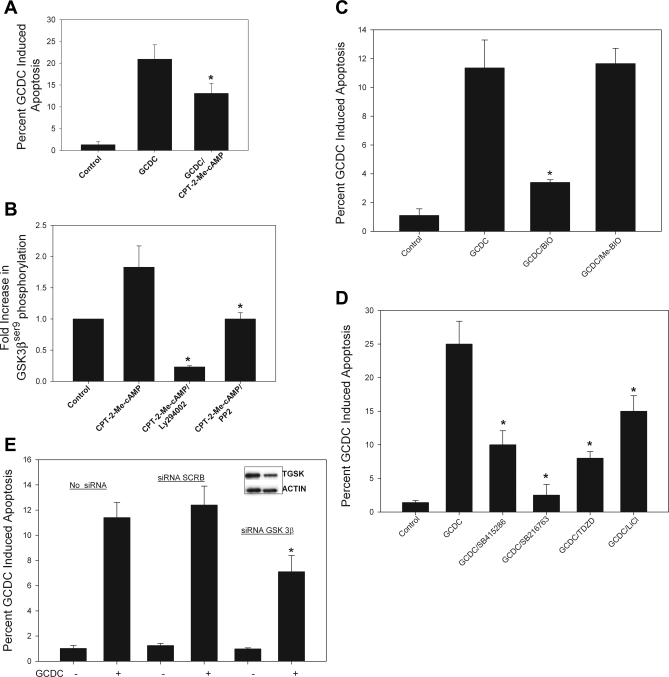
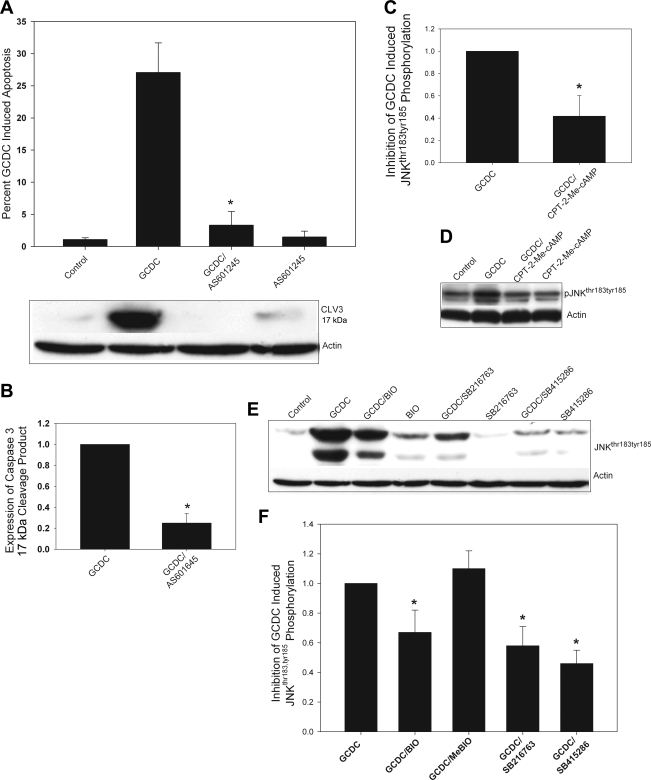
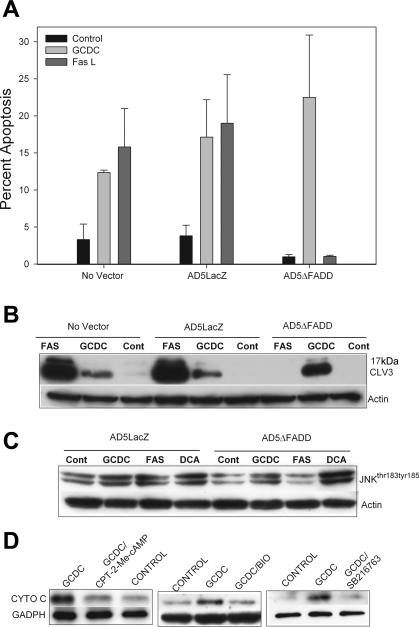
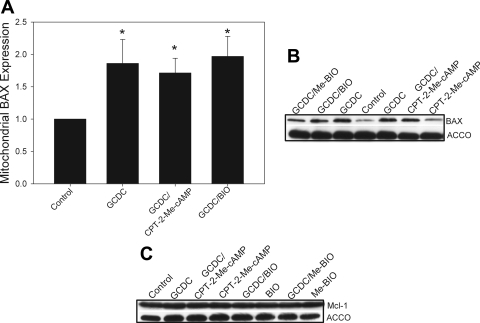
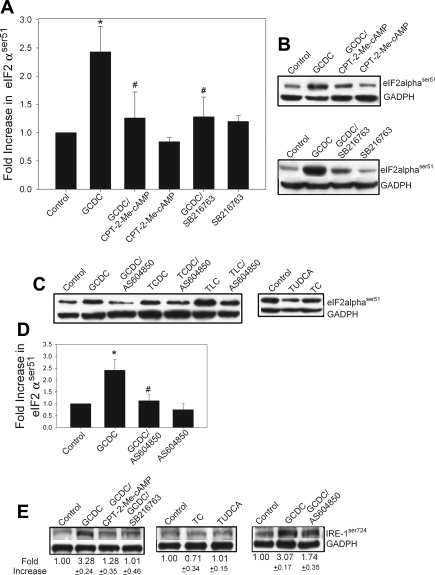
Cholestatic liver disorders are accompanied by the hepatic accumulation of cytotoxic bile acids that induce cell death. Increases in cAMP protect hepatocytes from bile acid-induced apoptosis by a cAMP-guanine exchange factor (cAMP-GEF)/phosphoinositide-3-kinase (PI3K)/Akt pathway. The aim of these studies was to identify the downstream substrate in this pathway and to determine at what level in the apoptotic cascade cytoprotection occurs. Since inhibitory phosphorylation of glycogen synthase kinase-3 (GSK) occurs downstream of PI3K/Akt and this phosphorylation has been implicated in cell survival, we conducted studies to determine whether GSK was downstream in cAMP-GEF/PI3K/Akt-mediated cytoprotection. Our results show that treatment of hepatocytes with the cAMP-GEF-specific analog, 4-(4-chlorophenylthio)-2'-O-methyladenosine-3',5'-cAMP, results in PI3K-dependent phosphorylation of GSK. Direct chemical inhibition of GSK in rat hepatocytes or human HUH7-NTCP cells with several structurally and functionally distinct inhibitors including bromoindirubin-3'-oxime (BIO), maleimides (SB216763, SB415286), thiadiazolidine derivatives, and LiCl attenuates apoptosis induced by glycochenodeoxycholate (GCDC). In addition, genetic silencing of the GSK β isoform with small interfering RNA attenuates GCDC apoptosis in HUH7-NTCP cells. Adenoviral inhibition of the Rap1 blocks both cAMP-GEF-mediated cytoprotection against GCDC-induced apoptosis and Akt/GSK3β phosphorylation. GCDC-induced phosphorylation of the proapoptotic kinase, c-Jun NH(2)-terminal kinase (JNK) is inhibited by GSK inhibition or cAMP-GEF activation. GCDC-induced apoptosis is accompanied by phosphorylation of the endoplasmic reticulum stress markers pIEF2α and IRE-1, and pretreatment with the cAMP-GEF analog or GSK inhibitors prevents this phosphorylation. Collectively, our results support the presence of a cAMP/cAMP-GEF/Rap1/PI3K/Akt/GSKβ survival pathway in hepatocytes that inhibits bile acid-induced JNK phosphorylation.
Figures
References
-
- Aghdam SY, Barger SW. Glycogen synthase kinase-3 in neurodegeneration and neuroprotection: lessons from lithium. Curr Alzheimer Res 4: 21–31, 2007 - PubMed
-
- Avila J, Wandosell F, Hernández F. Role of glycogen synthase kinase-3 in Alzheimer's disease: pathogenesis and glycogen kinase-3 inhibitors. Expert Rev Neurother 10: 703–710, 2010 - PubMed
-
- Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC. Involvement of cAMP/Epac/PI3K-dependent pathway in the antiproteolytic effect of epinephrine on rat skeletal muscle. Mol Cell Endrocrinol 315: 104–112, 2010 - PubMed
-
- Beauchard A, Ferandin Y, Frère S, Lozach O, Blairvacq M, Meijer L, Thiéry V, Besson T. Synthesis of novel 5-substituted indirubins as protein kinases. Bioorg Med Chem 14: 6434–6443, 2006 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous