

Comparative biology of cystic fibrosis animal models
- PMID: 21547741
- PMCID: PMC3617920
- DOI: 10.1007/978-1-61779-120-8_19
Comparative biology of cystic fibrosis animal models
Abstract
Animal models of human diseases are critical for dissecting mechanisms of pathophysiology and developing therapies. In the context of cystic fibrosis (CF), mouse models have been the dominant species by which to study CF disease processes in vivo for the past two decades. Although much has been learned through these CF mouse models, limitations in the ability of this species to recapitulate spontaneous lung disease and several other organ abnormalities seen in CF humans have created a need for additional species on which to study CF. To this end, pig and ferret CF models have been generated by somatic cell nuclear transfer and are currently being characterized. These new larger animal models have phenotypes that appear to closely resemble human CF disease seen in newborns, and efforts to characterize their adult phenotypes are ongoing. This chapter will review current knowledge about comparative lung cell biology and cystic fibrosis transmembrane conductance regulator (CFTR) biology among mice, pigs, and ferrets that has implications for CF disease modeling in these species. We will focus on methods used to compare the biology and function of CFTR between these species and their relevance to phenotypes seen in the animal models. These cross-species comparisons and the development of both the pig and the ferret CF models may help elucidate pathophysiologic mechanisms of CF lung disease and lead to new therapeutic approaches.
Figures
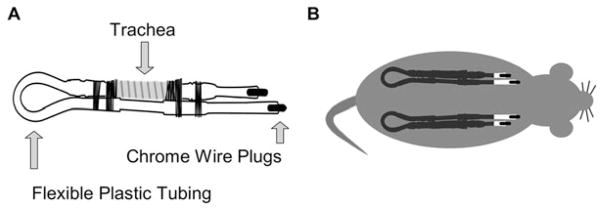
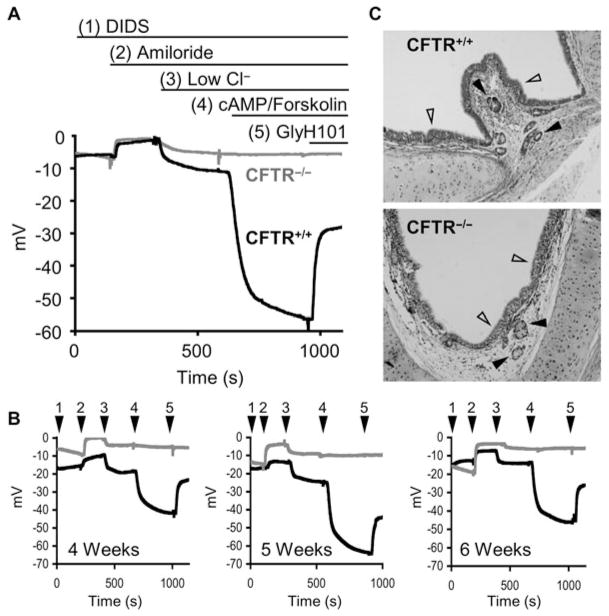
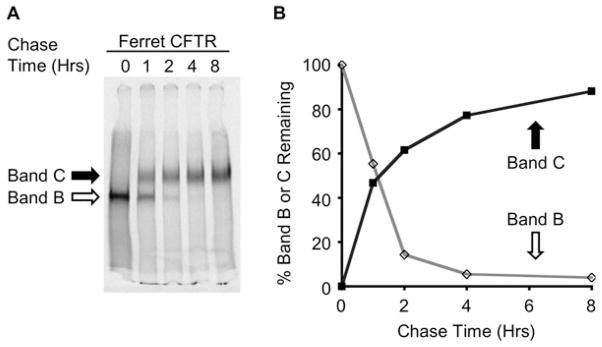
References
-
- Davidson DJ, Dorin JR. The CF mouse: an important tool for studying cystic fibrosis. Expert Rev Mol Med. 2001;3:1–27. - PubMed
-
- Davidson DJ, Rolfe M. Mouse models of cystic fibrosis. Trends Genet. 2001;17:S29–S37. - PubMed
-
- Dickinson P, Dorin JR, Porteous DJ. Modelling cystic fibrosis in the mouse. Mol Med Today. 1995;1:140–148. - PubMed
-
- Grubb BR, Boucher RC. Pathophysiology of gene-targeted mouse models for cystic fibrosis. Physiol Rev. 1999;79:S193–S214. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
