

Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP
- PMID: 21549340
- PMCID: PMC3146727
- DOI: 10.1016/j.ajhg.2011.04.002
Chondrodysplasia and abnormal joint development associated with mutations in IMPAD1, encoding the Golgi-resident nucleotide phosphatase, gPAPP
Abstract
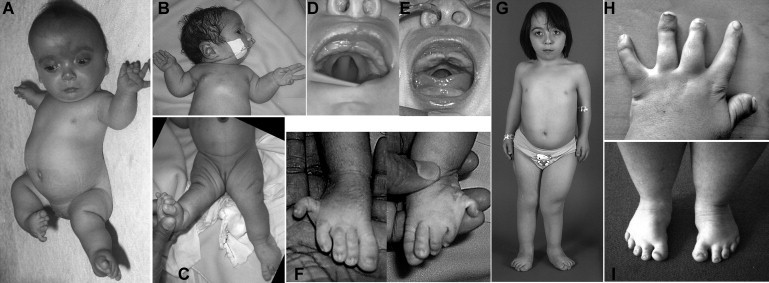
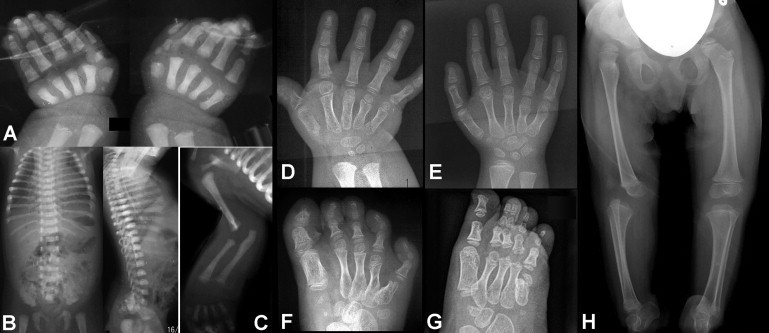
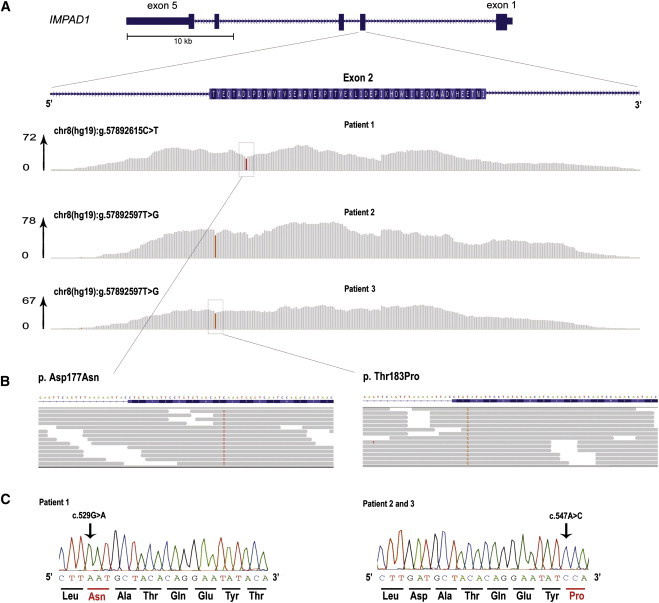
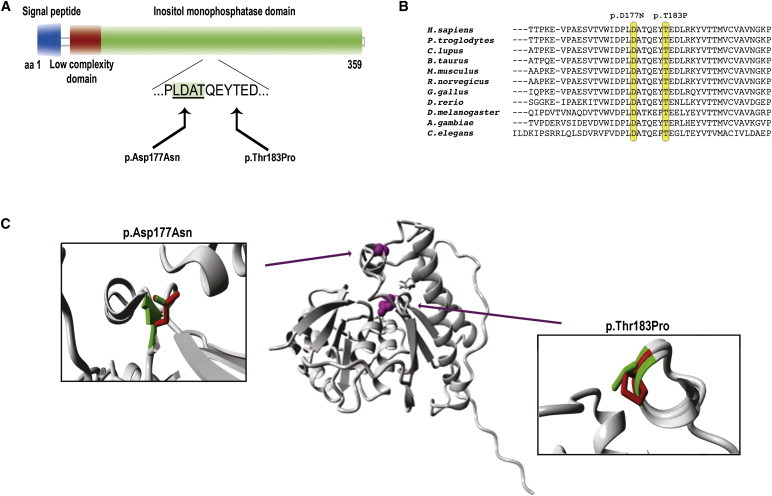
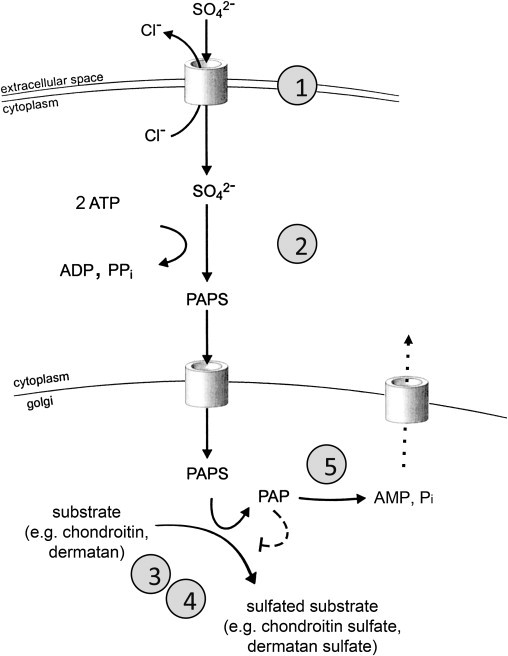
We used whole-exome sequencing to study three individuals with a distinct condition characterized by short stature, chondrodysplasia with brachydactyly, congenital joint dislocations, cleft palate, and facial dysmorphism. Affected individuals carried homozygous missense mutations in IMPAD1, the gene coding for gPAPP, a Golgi-resident nucleotide phosphatase that hydrolyzes phosphoadenosine phosphate (PAP), the byproduct of sulfotransferase reactions, to AMP. The mutations affected residues in or adjacent to the phosphatase active site and are predicted to impair enzyme activity. A fourth unrelated patient was subsequently found to be homozygous for a premature termination codon in IMPAD1. Impad1 inactivation in mice has previously been shown to produce chondrodysplasia with abnormal joint formation and impaired proteoglycan sulfation. The human chondrodysplasia associated with gPAPP deficiency joins a growing number of skeletoarticular conditions associated with defective synthesis of sulfated proteoglycans, highlighting the importance of proteoglycans in the development of skeletal elements and joints.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Krakow D., Robertson S.P., King L.M., Morgan T., Sebald E.T., Bertolotto C., Wachsmann-Hogiu S., Acuna D., Shapiro S.S., Takafuta T. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat. Genet. 2004;36:405–410. - PubMed
-
- Rossi A., Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum. Mutat. 2001;17:159–171. - PubMed
-
- Superti-Furga A. Defects in sulfate metabolism and skeletal dysplasias. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., Vogelstein B., Childs B., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; New York: 2001. pp. 5189–5201.
-
- Hermanns P., Unger S., Rossi A., Perez-Aytes A., Cortina H., Bonafé L., Boccone L., Setzu V., Dutoit M., Sangiorgi L. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am. J. Hum. Genet. 2008;82:1368–1374. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
