

Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy
- PMID: 21549344
- PMCID: PMC3146718
- DOI: 10.1016/j.ajhg.2011.04.006
Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy
Abstract
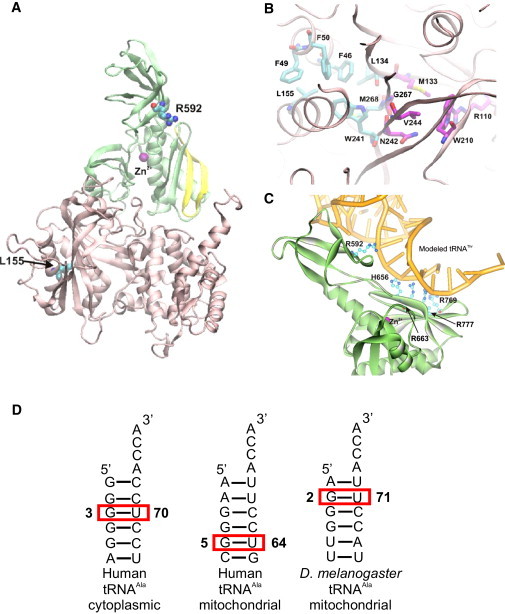
Infantile cardiomyopathies are devastating fatal disorders of the neonatal period or the first year of life. Mitochondrial dysfunction is a common cause of this group of diseases, but the underlying gene defects have been characterized in only a minority of cases, because tissue specificity of the manifestation hampers functional cloning and the heterogeneity of causative factors hinders collection of informative family materials. We sequenced the exome of a patient who died at the age of 10 months of hypertrophic mitochondrial cardiomyopathy with combined cardiac respiratory chain complex I and IV deficiency. Rigorous data analysis allowed us to identify a homozygous missense mutation in AARS2, which we showed to encode the mitochondrial alanyl-tRNA synthetase (mtAlaRS). Two siblings from another family, both of whom died perinatally of hypertrophic cardiomyopathy, had the same mutation, compound heterozygous with another missense mutation. Protein structure modeling of mtAlaRS suggested that one of the mutations affected a unique tRNA recognition site in the editing domain, leading to incorrect tRNA aminoacylation, whereas the second mutation severely disturbed the catalytic function, preventing tRNA aminoacylation. We show here that mutations in AARS2 cause perinatal or infantile cardiomyopathy with near-total combined mitochondrial respiratory chain deficiency in the heart. Our results indicate that exome sequencing is a powerful tool for identifying mutations in single patients and allows recognition of the genetic background in single-gene disorders of variable clinical manifestation and tissue-specific disease. Furthermore, we show that mitochondrial disorders extend to prenatal life and are an important cause of early infantile cardiac failure.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures




References
-
- Thorburn D.R. Mitochondrial disorders: prevalence, myths and advances. J. Inherit. Metab. Dis. 2004;27:349–362. - PubMed
-
- Holmgren D., Wåhlander H., Eriksson B.O., Oldfors A., Holme E., Tulinius M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur. Heart J. 2003;24:280–288. - PubMed
-
- Scaglia F., Towbin J.A., Craigen W.J., Belmont J.W., Smith E.O., Neish S.R., Ware S.M., Hunter J.V., Fernbach S.D., Vladutiu G.D. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925–931. - PubMed
-
- Yaplito-Lee J., Weintraub R., Jamsen K., Chow C.W., Thorburn D.R., Boneh A. Cardiac manifestations in oxidative phosphorylation disorders of childhood. J. Pediatr. 2007;150:407–411. - PubMed
-
- Tanaka M., Ino H., Ohno K., Hattori K., Sato W., Ozawa T., Tanaka T., Itoyama S. Mitochondrial mutation in fatal infantile cardiomyopathy. Lancet. 1990;336:1452. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
