

Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes
- PMID: 21551504
- PMCID: PMC3149096
- DOI: 10.1074/mcp.M111.009654
Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes
Abstract
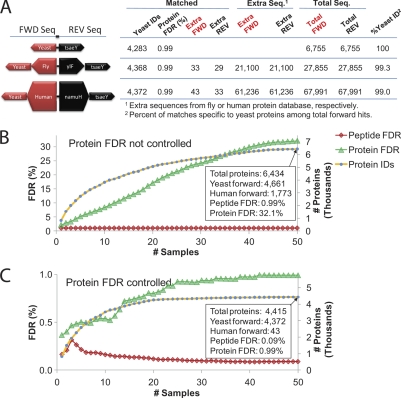
The interpretation of quantitative phosphoproteomics studies is complicated because each differential phosphorylation event integrates both changes in protein expression and phosphorylation. Here we investigated this phenomenon by performing parallel comparisons of protein expression and phosphorylation in S. cerevisiae. In each of two experiments comparing yeast mutants bearing deletions in FUS3 or STE7 with their wild-type counterparts, we quantified over 4100 proteins, including all members of the yeast mating pathway. We also identified 12,499 unique phosphorylation sites in this work. We demonstrate the critical importance of controlling the protein-level false-discovery rate and provide a novel method to assess the accuracy of protein false-discovery rate estimates. For the first time, 96% of nonredundant phosphopeptide ratios could be calibrated by protein levels, allowing truly differential phosphorylation to be distinguished from altered protein expression. This revealed a starkly different view, with 25% of seemingly differential phosphopeptides now attributed to changes in protein expression. Combined protein expression and phosphorylation surveys uncovered both independent and concerted changes in protein expression and phosphorylation, while highlighting the partially redundant role of a second MAPK (Kss1) in the mating pathway.
Figures





References
-
- Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. - PubMed
-
- Choudhary C., Olsen J. V., Brandts C., Cox J., Reddy P. N., Böhmer F. D., Gerke V., Schmidt-Arras D. E., Berdel W. E., Müller-Tidow C., Mann M., Serve H. (2009) Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell. 36, 326–339 - PubMed
-
- Wiśniewski J. R., Nagaraj N., Zougman A., Gnad F., Mann M. (2010) Brain Phosphoproteome Obtained by a FASP-Based Method Reveals Plasma Membrane Protein Topology. J. Proteome. Res. 9, 3280–3289 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
