

KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes
- PMID: 21552264
- PMCID: PMC3674836
- DOI: 10.1038/ng.826
KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes
Abstract
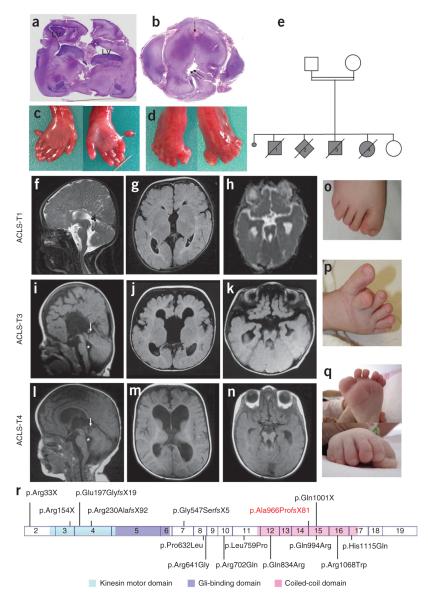
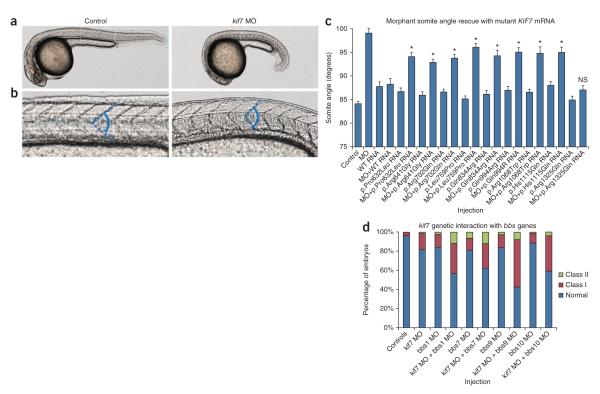
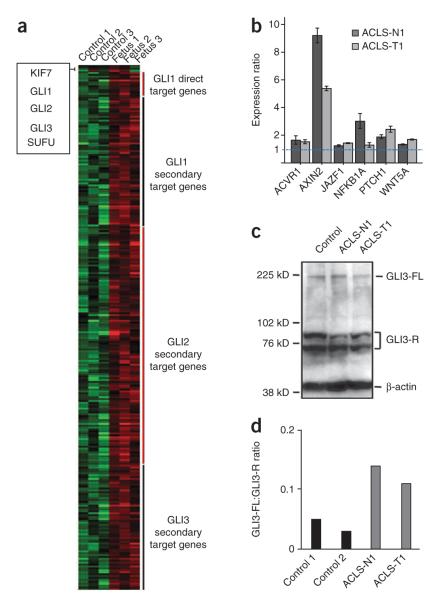
KIF7, the human ortholog of Drosophila Costal2, is a key component of the Hedgehog signaling pathway. Here we report mutations in KIF7 in individuals with hydrolethalus and acrocallosal syndromes, two multiple malformation disorders with overlapping features that include polydactyly, brain abnormalities and cleft palate. Consistent with a role of KIF7 in Hedgehog signaling, we show deregulation of most GLI transcription factor targets and impaired GLI3 processing in tissues from individuals with KIF7 mutations. KIF7 is also a likely contributor of alleles across the ciliopathy spectrum, as sequencing of a diverse cohort identified several missense mutations detrimental to protein function. In addition, in vivo genetic interaction studies indicated that knockdown of KIF7 could exacerbate the phenotype induced by knockdown of other ciliopathy transcripts. Our data show the role of KIF7 in human primary cilia, especially in the Hedgehog pathway through the regulation of GLI targets, and expand the clinical spectrum of ciliopathies.
Figures
References
-
- Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:281–295. - PubMed
-
- Salonen R, Herva R, Norio R. The hydrolethalus syndrome: delineation of a “new,” lethal malformation syndrome based on 28 patients. Clin. Genet. 1981;19:321–330. - PubMed
-
- Mee L, et al. Hydrolethalus syndrome is caused by a missense mutation in a novel gene HYLS1. Hum. Mol. Genet. 2005;14:1475–1488. - PubMed
-
- Schinzel A, Schmid W. Hallux duplication, postaxial polydactyly, absence of the corpus callosum, severe mental retardation, and additional anomalies in two unrelated patients: a new syndrome. Am. J. Med. Genet. 1980;6:241–249. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
