

Toxin models of mitochondrial dysfunction in Parkinson's disease
- PMID: 21554057
- PMCID: PMC3292753
- DOI: 10.1089/ars.2011.4033
Toxin models of mitochondrial dysfunction in Parkinson's disease
Abstract
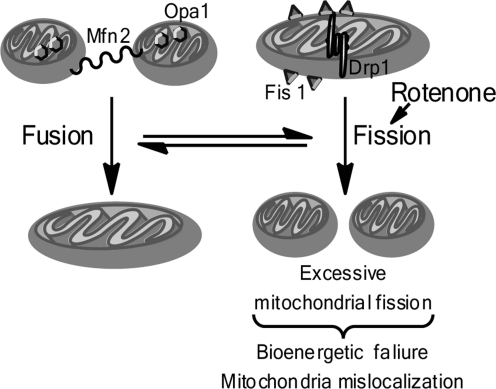
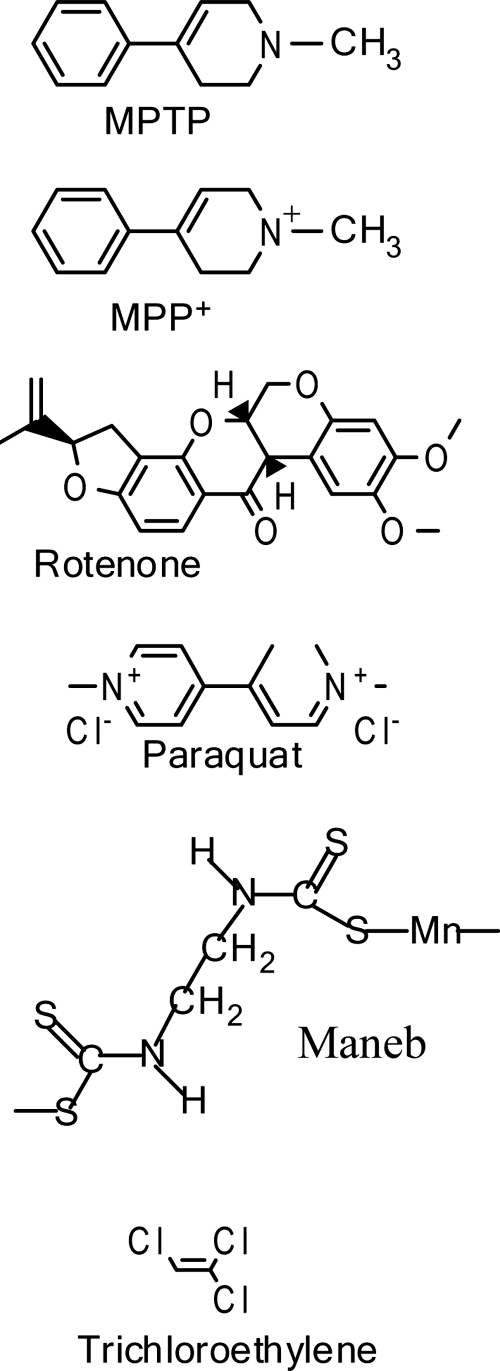
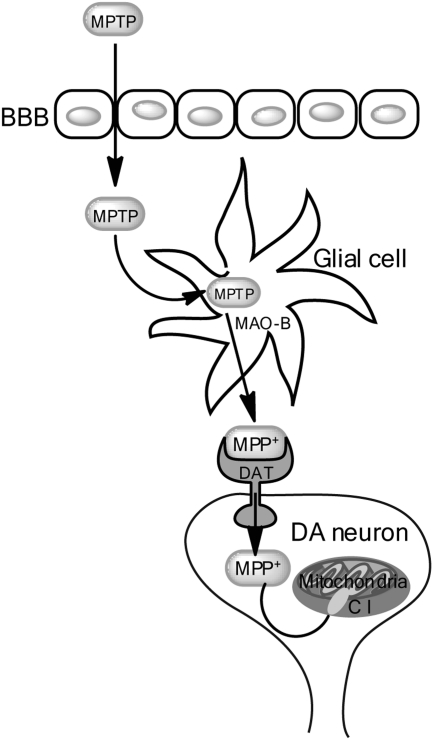
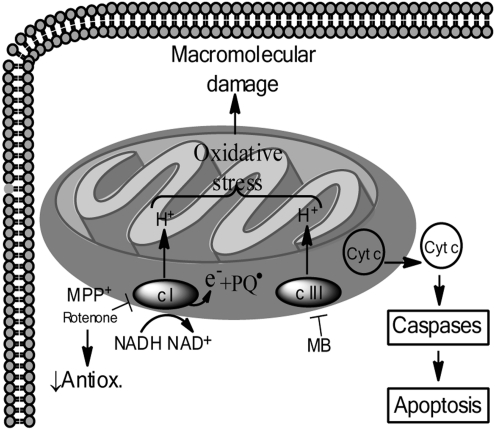
Significance: Parkinson's disease (PD) is a neurodegenerative disorder characterized, in part, by the progressive and selective loss of dopaminergic neuron cell bodies within the substantia nigra pars compacta (SNpc) and the associated deficiency of the neurotransmitter dopamine (DA) in the striatum, which gives rise to the typical motor symptoms of PD. The mechanisms that contribute to the induction and progressive cell death of dopaminergic neurons in PD are multi-faceted and remain incompletely understood. Data from epidemiological studies in humans and molecular studies in genetic, as well as toxin-induced animal models of parkinsonism, indicate that mitochondrial dysfunction occurs early in the pathogenesis of both familial and idiopathic PD. In this review, we provide an overview of toxin models of mitochondrial dysfunction in experimental Parkinson's disease and discuss mitochondrial mechanisms of neurotoxicity.
Recent advances: A new toxin model using the mitochondrial toxin trichloroethylene was recently described and novel methods, such as intranasal exposure to toxins, have been explored. Additionally, recent research conducted in toxin models of parkinsonism provides an emerging emphasis on extranigral aspects of PD pathology.
Critical issues: Unfortunately, none of the existing animal models of experimental PD completely mimics the etiology, progression, and pathology of human PD.
Future directions: Continued efforts to optimize established animal models of parkinsonism, as well as the development and characterization of new animal models are essential, as there still remains a disconnect in terms of translating mechanistic observations in animal models of experimental PD into bona fide disease-modifying therapeutics for human PD patients.
Figures
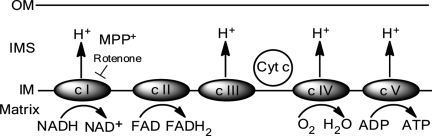
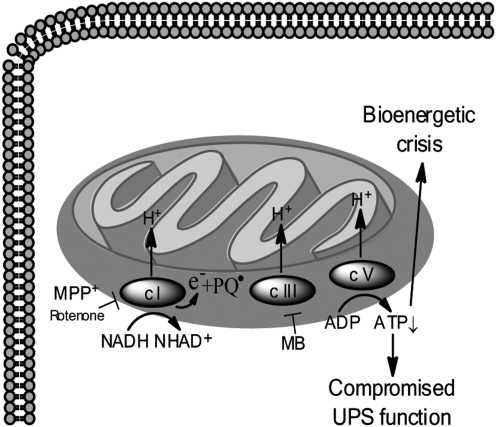
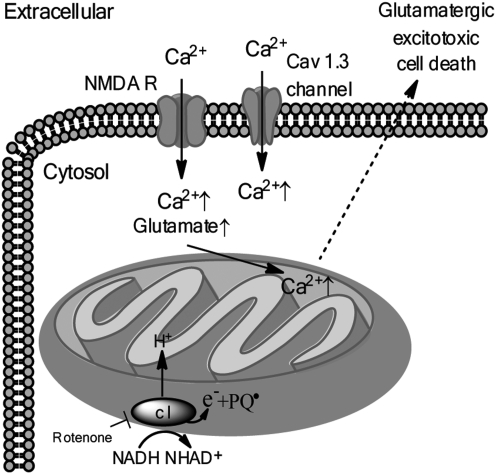
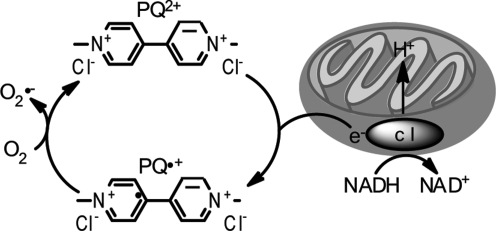
References
-
- Ahmadi FA. Linseman DA. Grammatopoulos TN. Jones SM. Bouchard RJ. Freed CR. Heidenreich KA. Zawada WM. The pesticide rotenone induces caspase-3-mediated apoptosis in ventral mesencephalic dopaminergic neurons. J Neurochem. 2003;87:914–921. - PubMed
-
- Ascherio A. Chen H. Weisskopf MG. O'Reilly E. McCullough ML. Calle EE. Schwarzschild MA. Thun MJ. Pesticide exposure and risk for Parkinson's disease. Ann Neurol. 2006;60:197–203. - PubMed
-
- Barrot M. Calza L. Pozza M. Le Moal M. Piazza PV. Differential calbindin-immunoreactivity in dopamine neurons projecting to the rat striatal complex. Eur J Neurosci. 2000;12:4578–4582. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
