

MetaPath: identifying differentially abundant metabolic pathways in metagenomic datasets
- PMID: 21554767
- PMCID: PMC3090767
- DOI: 10.1186/1753-6561-5-S2-S9
MetaPath: identifying differentially abundant metabolic pathways in metagenomic datasets
Abstract
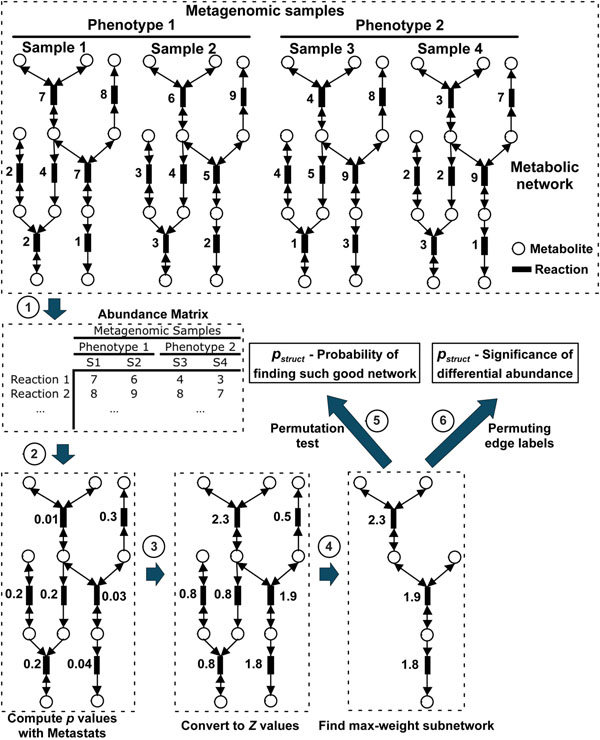
Background: Enabled by rapid advances in sequencing technology, metagenomic studies aim to characterize entire communities of microbes bypassing the need for culturing individual bacterial members. One major goal of metagenomic studies is to identify specific functional adaptations of microbial communities to their habitats. The functional profile and the abundances for a sample can be estimated by mapping metagenomic sequences to the global metabolic network consisting of thousands of molecular reactions. Here we describe a powerful analytical method (MetaPath) that can identify differentially abundant pathways in metagenomic datasets, relying on a combination of metagenomic sequence data and prior metabolic pathway knowledge.
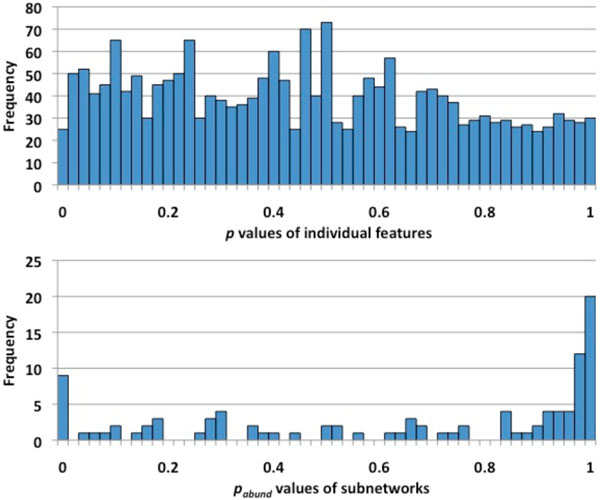
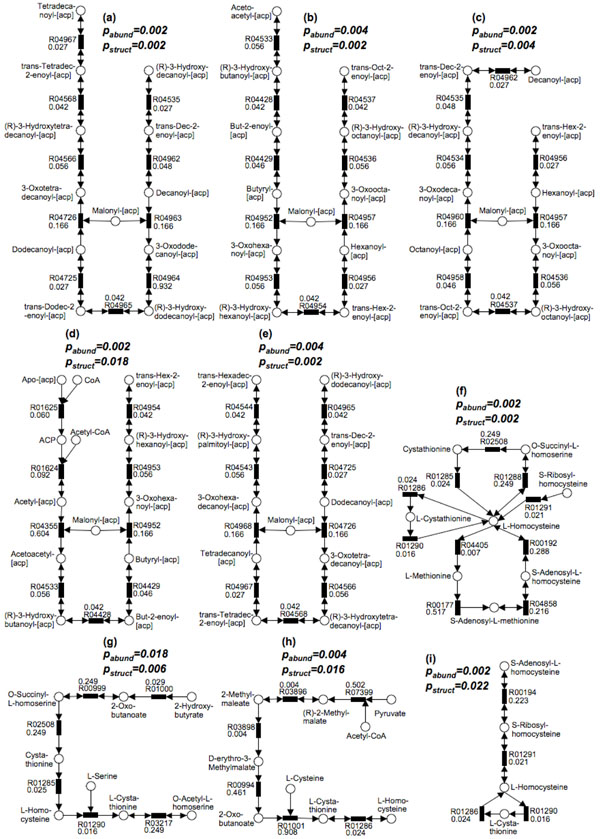
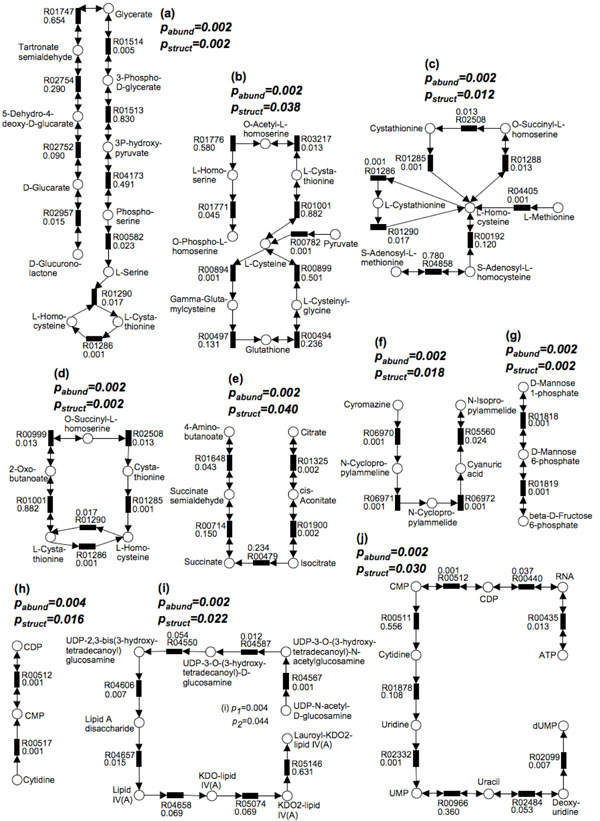
Methods: First, we introduce a scoring function for an arbitrary subnetwork and find the max-weight subnetwork in the global network by a greedy search algorithm. Then we compute two p values (pabund and pstruct) using nonparametric approaches to answer two different statistical questions: (1) is this subnetwork differentically abundant? (2) What is the probability of finding such good subnetworks by chance given the data and network structure? Finally, significant metabolic subnetworks are discovered based on these two p values.
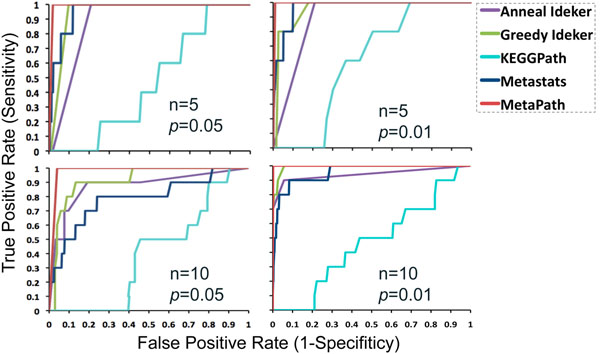
Results: In order to validate our methods, we have designed a simulated metabolic pathways dataset and show that MetaPath outperforms other commonly used approaches. We also demonstrate the power of our methods in analyzing two publicly available metagenomic datasets, and show that the subnetworks identified by MetaPath provide valuable insights into the biological activities of the microbiome.
Conclusions: We have introduced a statistical method for finding significant metabolic subnetworks from metagenomic datasets. Compared with previous methods, results from MetaPath are more robust against noise in the data, and have significantly higher sensitivity and specificity (when tested on simulated datasets). When applied to two publicly available metagenomic datasets, the output of MetaPath is consistent with previous observations and also provides several new insights into the metabolic activity of the gut microbiome. The software is freely available at http://metapath.cbcb.umd.edu.
Figures
References
-
- Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A. et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
