

Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease
- PMID: 21555590
- PMCID: PMC3102344
- DOI: 10.1073/pnas.1101964108
Hypomorphic Notch 3 alleles link Notch signaling to ischemic cerebral small-vessel disease
Abstract
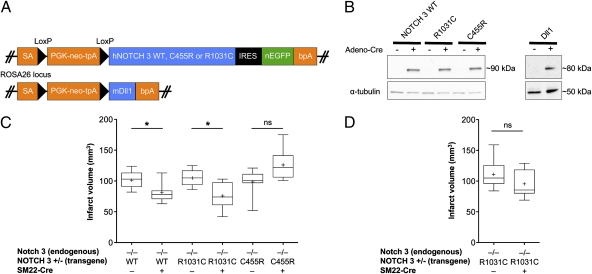
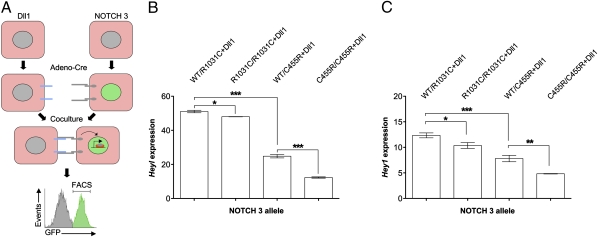
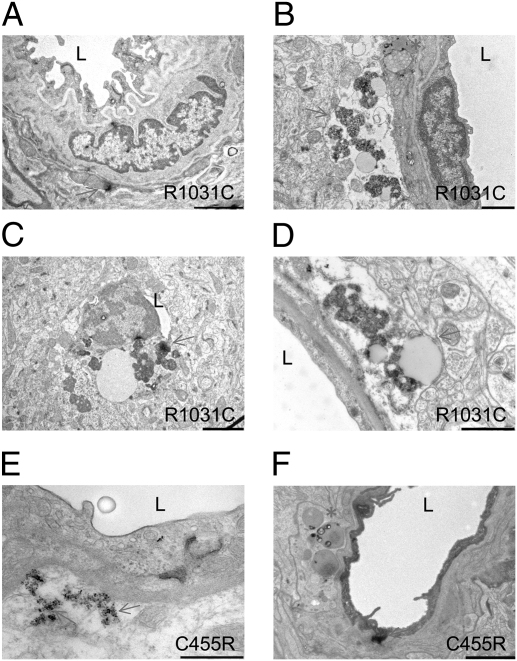
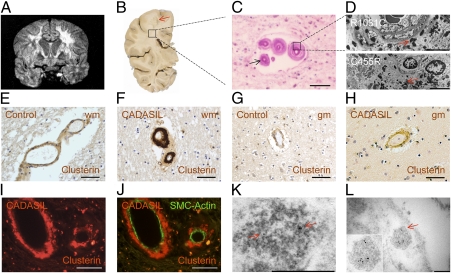
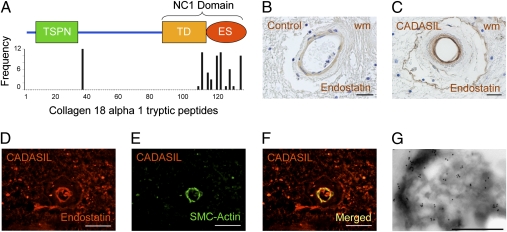
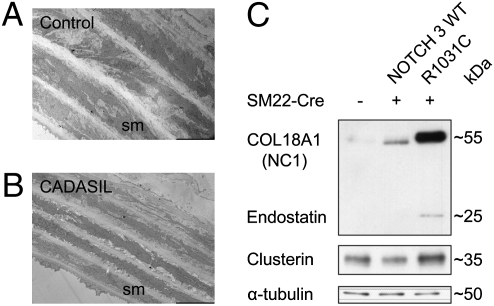
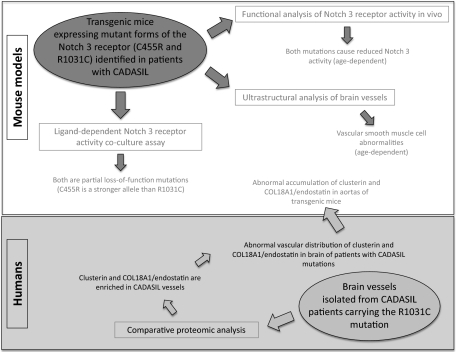
The most common monogenic cause of small-vessel disease leading to ischemic stroke and vascular dementia is the neurodegenerative syndrome cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), which is associated with mutations in the Notch 3 receptor. CADASIL pathology is characterized by vascular smooth muscle cell degeneration and accumulation of diagnostic granular osmiophilic material (GOM) in vessels. The functional nature of the Notch 3 mutations causing CADASIL and their mechanistic connection to small-vessel disease and GOM accumulation remain enigmatic. To gain insight into how Notch 3 function is linked to CADASIL pathophysiology, we studied two phenotypically distinct mutations, C455R and R1031C, respectively associated with early and late onset of stroke, by using hemodynamic analyses in transgenic mouse models, receptor activity assays in cell culture, and proteomic examination of postmortem human tissue. We demonstrate that the C455R and R1031C mutations define different hypomorphic activity states of Notch 3, a property linked to ischemic stroke susceptibility in mouse models we generated. Importantly, these mice develop osmiophilic deposits and other age-dependent phenotypes that parallel remarkably the human condition. Proteomic analysis of human brain vessels, carrying the same CADASIL mutations, identified clusterin and collagen 18 α1/endostatin as GOM components. Our findings link loss of Notch signaling with ischemic cerebral small-vessel disease, a prevalent human condition. We determine that CADASIL pathophysiology is associated with hypomorphic Notch 3 function in vascular smooth muscle cells and implicate the accumulation of clusterin and collagen 18 α1/endostatin in brain vessel pathology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Thompson CS, Hakim AM. Living beyond our physiological means: Small vessel disease of the brain is an expression of a systemic failure in arteriolar function: A unifying hypothesis. Stroke. 2009;40:e322–e330. - PubMed
-
- Joutel A, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. - PubMed
-
- Gould DB, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354:1489–1496. - PubMed
-
- Revesz T, et al. Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003;62:885–898. - PubMed
-
- Hara K, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360:1729–1739. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
