

SCA15 due to large ITPR1 deletions in a cohort of 333 white families with dominant ataxia
- PMID: 21555639
- PMCID: PMC3142680
- DOI: 10.1001/archneurol.2011.81
SCA15 due to large ITPR1 deletions in a cohort of 333 white families with dominant ataxia
Abstract
Background: Deletions in ITPR1, coding for the inositol-triphosphate receptor type 1, have been recently identified in spinocerebellar ataxia type 15 (SCA15).
Objective: To determine the frequency and the phenotypical spectrum of SCA15.
Design: Taqman polymerase chain reaction (258 index cases) or single-nucleotide polymorphism genome-wide genotyping (75 index cases).
Setting: A collaboration between the Centre de Recherche de l'Institut de Cerveau et de la Moelle Epinière of the Salpêtrière Hospital (Paris, France) and the Molecular Genetics Unit of the National Institute of Aging (Bethesda, Maryland). Patients Index cases of 333 families with autosomal dominant cerebellar ataxia negative for CAG repeat expansions in coding exons.
Main outcome measures: Detection of ITPR1 copy number alterations.
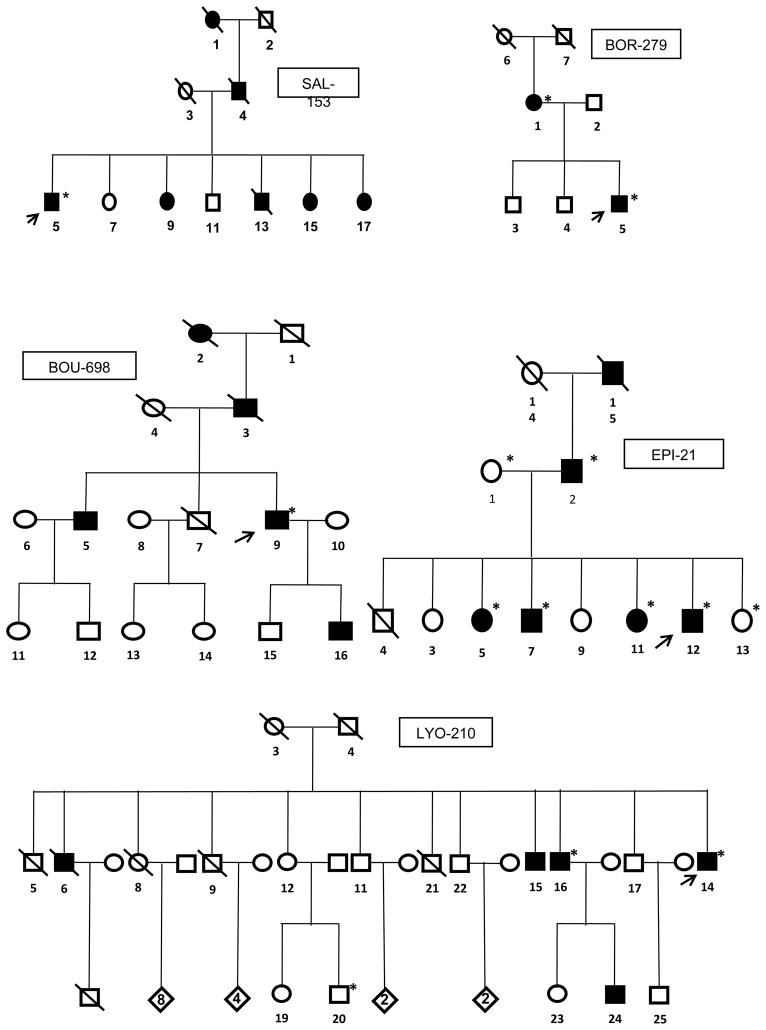
Results: A deletion of ITPR1 was found in 6 of 333 families (1.8%), corresponding to 13 patients with SCA15. Age at onset ranged from 18 to 66 years (mean [SD] age, 35 [16] years). The symptom at onset was cerebellar gait ataxia, except in 1 patient with isolated upper limb tremor. Although families were tested irrespective of their phenotype, patients with SCA15 had a homogeneous phenotype and were characterized by a slowly progressive cerebellar ataxia. However, pyramidal signs (2 patients) and mild cognitive problems (2 patients) were occasionally present. Radiologic findings showed global or predominant vermian cerebellar atrophy in all patients.
Conclusions: In this series, ITPR1 deletions were rare and accounted for approximately 1% of all autosomal dominant cerebellar ataxias. The SCA15 phenotype mostly consists of a slowly progressive isolated cerebellar ataxia with variable age at onset; an additional pyramidal syndrome and problems in executive functions may be present.
Figures


References
-
- Stevanin G, Dürr A, Brice A. Spinocerebellar ataxias caused by polyglutamine expansions. Adv Exp Med Biol. 2002;516:47–77. - PubMed
-
- Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3(5):291–304. - PubMed
-
- Stevanin G, Dürr A, Brice A. Clinical and genetic aspects of spinocerebellar ataxias with emphasis on polyglutamine expansions. Spinocerebellar degenerations: the ataxias and spastic paraplegias. In: Brice A, Pulst S, editors. Blue books of Neurology. Vol. 31. Elsevier; 2007. pp. 113–144.
-
- Brice A, Pulst S, editors. Blue books of Neurology. Elsevier; 2007. Spinocerebellar degenerations: the ataxias and spastic paraplegias.
-
- Dürr A, Stevanin G, Forlani S, et al. Conventional mutations are associated with a different phenotype than polyglutamine expansions in spinocerebellar ataxias. Eur J Human Gen. 2009;17(suppl 2):p335. Abstract.
