

The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice
- PMID: 21555854
- PMCID: PMC3104772
- DOI: 10.1172/JCI46277
The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice
Abstract
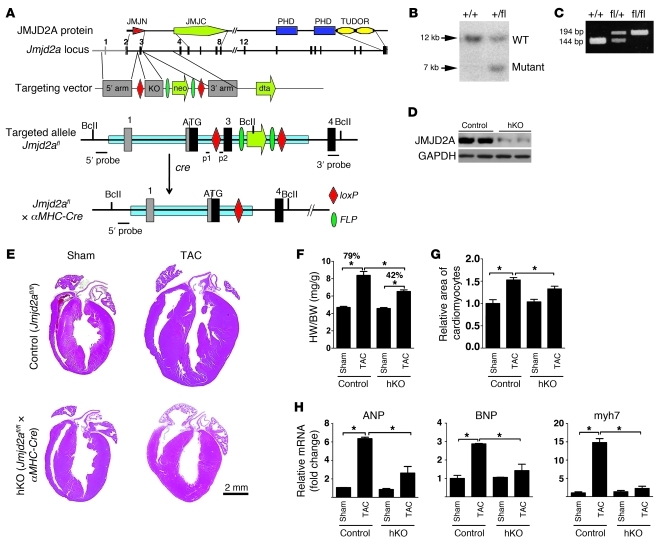
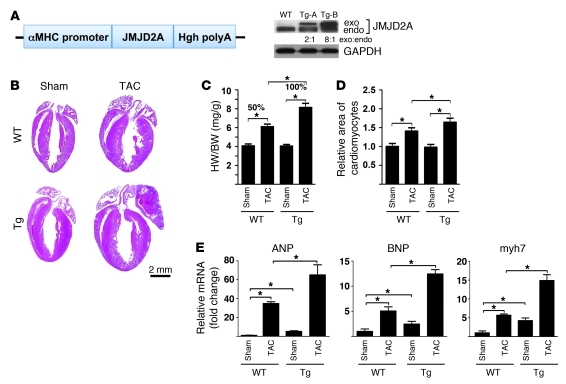
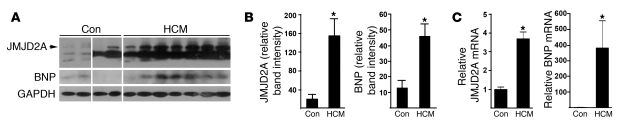
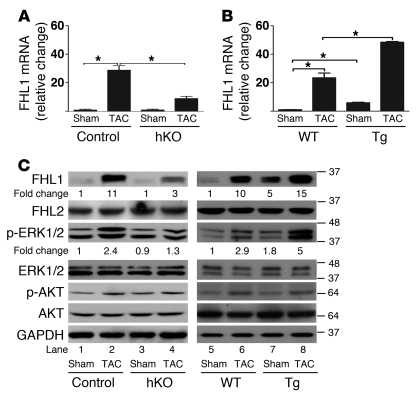
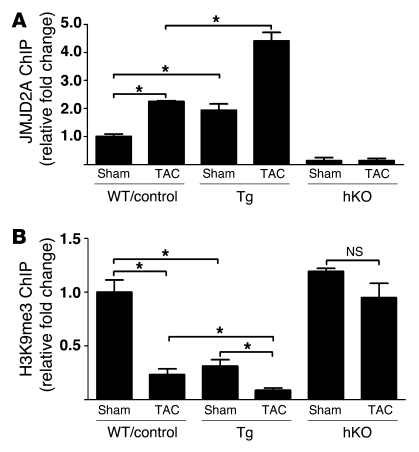
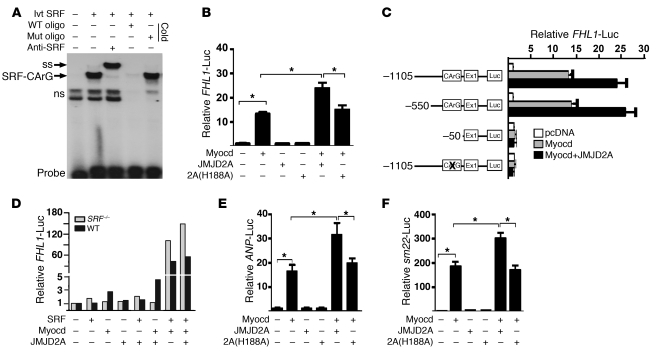
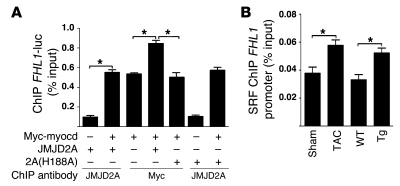
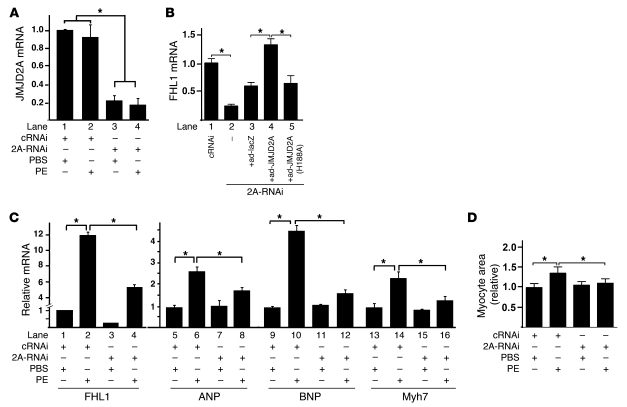
Cardiac hypertrophy and failure are accompanied by a reprogramming of gene expression that involves transcription factors and chromatin remodeling enzymes. Little is known about the roles of histone methylation and demethylation in this process. To understand the role of JMJD2A, a histone trimethyl demethylase, in cardiac hypertrophy, we generated mouse lines with heart-specific Jmjd2a deletion (hKO) and overexpression (Jmjd2a-Tg). Jmjd2a hKO and Jmjd2a-Tg mice had no overt baseline phenotype, but did demonstrate altered responses to cardiac stresses. While inactivation of Jmjd2a resulted in an attenuated hypertrophic response to transverse aortic constriction-induced (TAC-induced) pressure overload, Jmjd2a-Tg mice displayed exacerbated cardiac hypertrophy. We identified four-and-a-half LIM domains 1 (FHL1), a key component of the mechanotransducer machinery in the heart, as a direct target of JMJD2A. JMJD2A bound to the FHL1 promoter in response to TAC, upregulated FHL1 expression, and downregulated H3K9 trimethylation. Upregulation of FHL1 by JMJD2A was mediated through SRF and myocardin and required its demethylase activity. The expression of JMJD2A was upregulated in human hypertrophic cardiomyopathy patients. Our studies reveal that JMJD2A promotes cardiac hypertrophy under pathological conditions and suggest what we believe to be a novel mechanism for JMJD2A in reprogramming of gene expression involved in cardiac hypertrophy.
Figures
References
-
- Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322(22):1561–1566. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL-080144/HL/NHLBI NIH HHS/United States
- F32 HL009010/HL/NHLBI NIH HHS/United States
- HL-090842/HL/NHLBI NIH HHS/United States
- R01 HL109471/HL/NHLBI NIH HHS/United States
- T32 HL007360-31/HL/NHLBI NIH HHS/United States
- R01 HL080144/HL/NHLBI NIH HHS/United States
- HL-075173/HL/NHLBI NIH HHS/United States
- R01 HL090842/HL/NHLBI NIH HHS/United States
- R01 HL075173/HL/NHLBI NIH HHS/United States
- U01-RFA-HL09-010/HL/NHLBI NIH HHS/United States
- R01 HL085749/HL/NHLBI NIH HHS/United States
- T32 HL007360/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
