

Chemical treatment enhances skipping of a mutated exon in the dystrophin gene
- PMID: 21556062
- PMCID: PMC3113229
- DOI: 10.1038/ncomms1306
Chemical treatment enhances skipping of a mutated exon in the dystrophin gene
Abstract
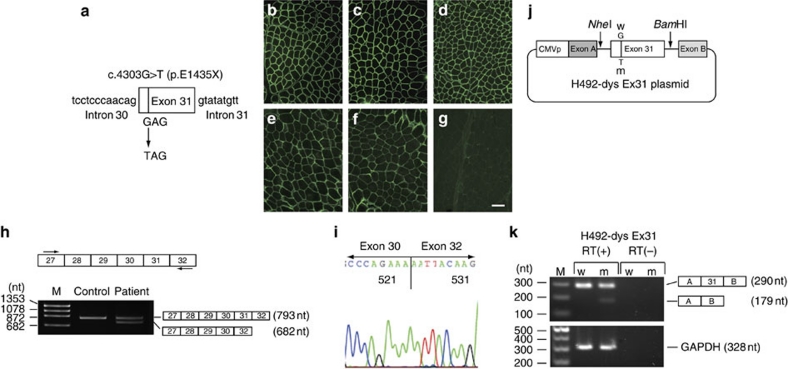
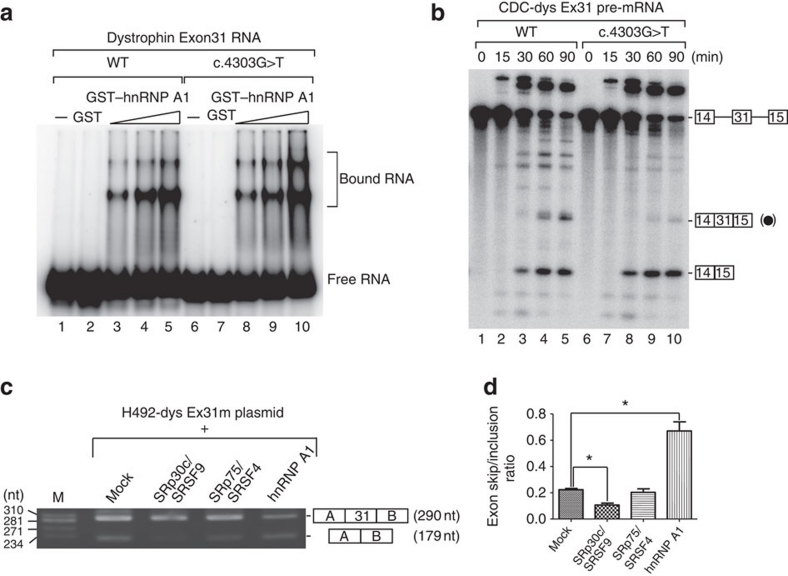
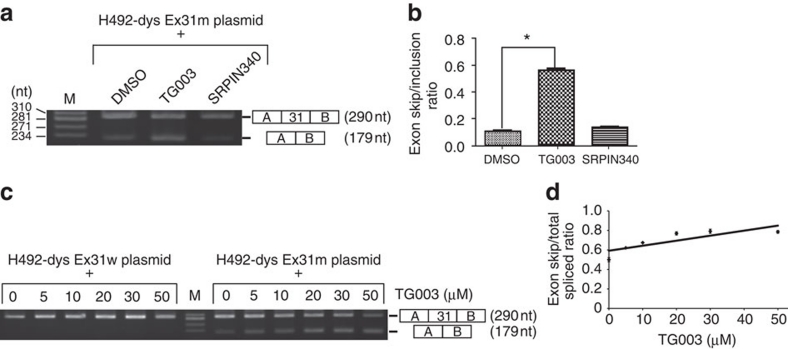
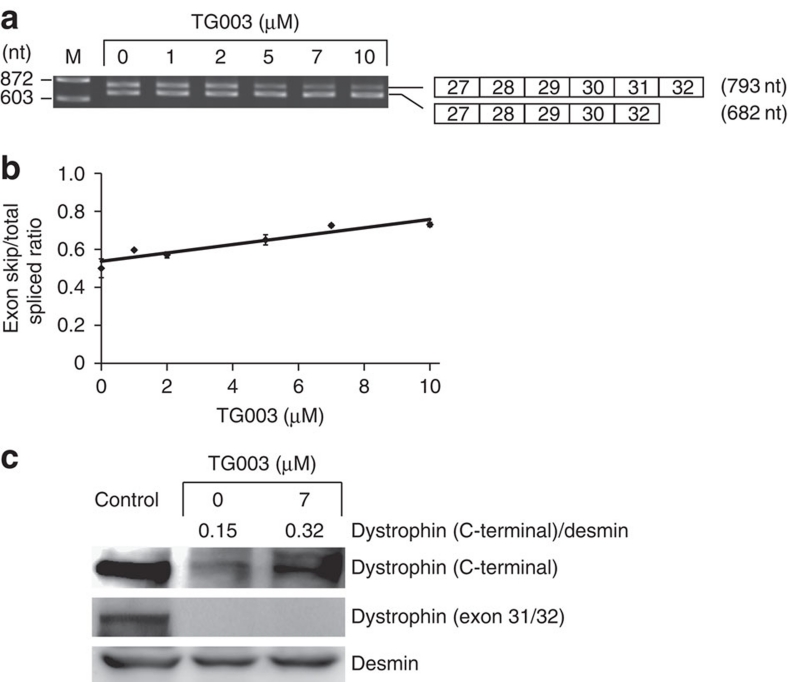
Duchenne muscular dystrophy (DMD) is a fatal muscle wasting disease caused by a loss of the dystrophin protein. Control of dystrophin mRNA splicing to convert severe DMD to a milder phenotype is attracting much attention. Here we report a dystrophinopathy patient who has a point mutation in exon 31 of the dystrophin gene. Although the mutation generates a stop codon, a small amount of internally deleted, but functional, dystrophin protein is produced in the patient cells. An analysis of the mRNA reveals that the mutation promotes exon skipping and restores the open reading frame of dystrophin. Presumably, the mutation disrupts an exonic splicing enhancer and creates an exonic splicing silencer. Therefore, we searched for small chemicals that enhance exon skipping, and found that TG003 promotes the skipping of exon 31 in the endogenous dystrophin gene in a dose-dependent manner and increases the production of the dystrophin protein in the patient's cells.
Figures
References
-
- Koenig M., Monaco A. P. & Kunkel L. M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 53, 219–228 (1988). - PubMed
-
- Monaco A. P., Bertelson C. J., Liechti-Gallati S., Moser H. & Kunkel L. M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1988). - PubMed
-
- Nishiyama A. et al. Dystrophin nonsense mutations can generate alternative rescue transcripts in lymphocytes. Ann. Hum. Genet. 72, 717–724 (2008). - PubMed
-
- Disset A. et al. An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum. Mol. Genet. 15, 999–1013 (2006). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
