

Translational research and therapeutic perspectives in dysferlinopathies
- PMID: 21556485
- PMCID: PMC3188867
- DOI: 10.2119/molmed.2011.00084
Translational research and therapeutic perspectives in dysferlinopathies
Abstract
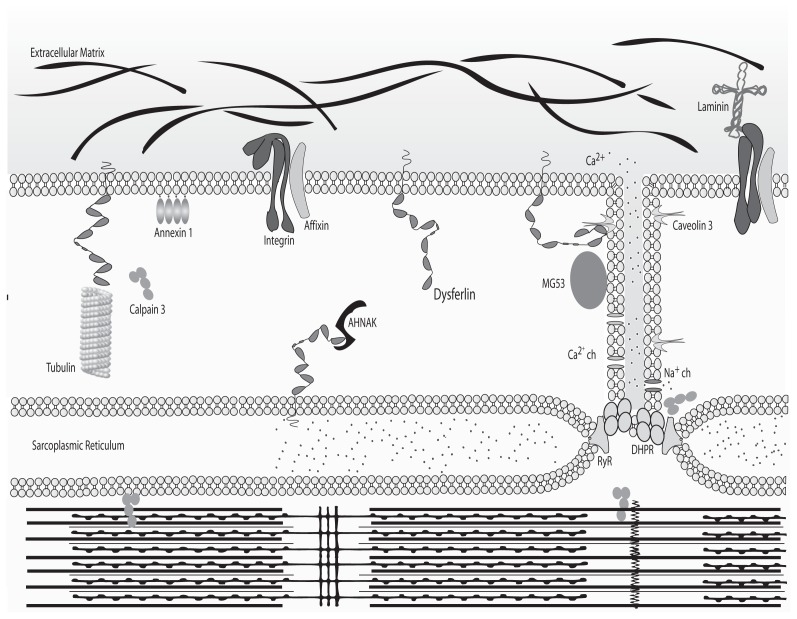
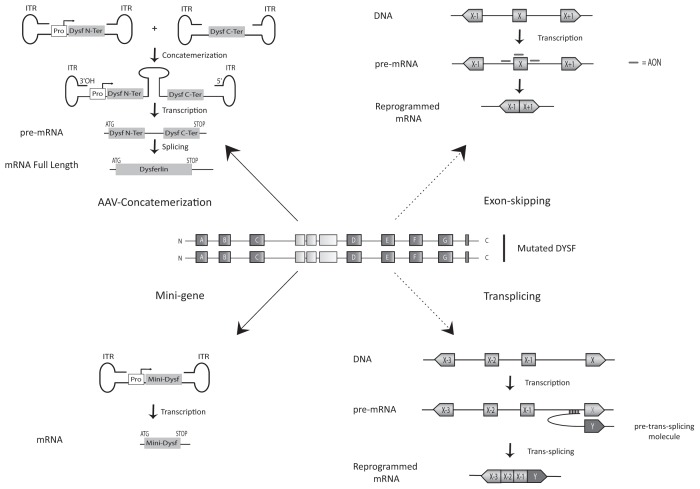
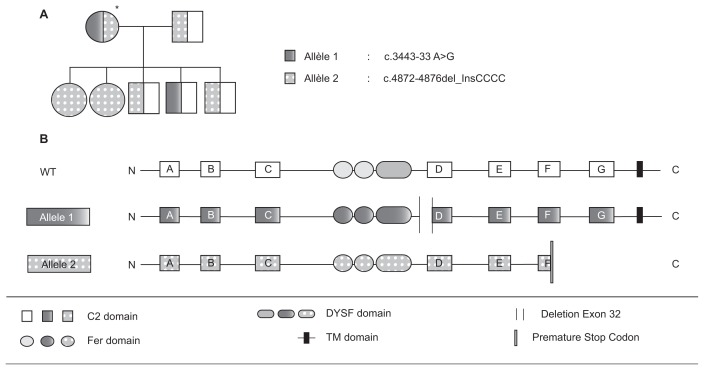
Dysferlinopathies are autosomal recessive disorders caused by mutations in the dysferlin (DYSF) gene, encoding the dysferlin protein. DYSF mutations lead to a wide range of muscular phenotypes, with the most prominent being Miyoshi myopathy (MM) and limb girdle muscular dystrophy type 2B (LGMD2B) and the second most common being LGMD. Symptoms generally appear at the end of childhood and, although disease progression is typically slow, walking impairments eventually result. Dysferlin is a modular type II transmembrane protein for which numerous binding partners have been identified. Although dysferlin function is only partially elucidated, this large protein contains seven calcium sensor C2 domains, shown to play a key role in muscle membrane repair. On the basis of this major function, along with detailed clinical observations, it has been possible to design various therapeutic approaches for dysferlin-deficient patients. Among them, exon-skipping and minigene transfer strategies have been evaluated at the preclinical level and, to date, represent promising approaches for clinical trials. This review aims to summarize the pathophysiology of dysferlinopathies and to evaluate the therapeutic potential for treatments currently under development.
Figures
References
-
- Bashir R, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20:37–42. - PubMed
-
- Liu J, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–6. - PubMed
-
- Moore SA, et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol. 2006;65:995–1003. - PubMed
-
- Nguyen K, et al. Phenotypic study in 40 patients with dysferlin gene mutations: high frequency of atypical phenotypes. Arch Neurol. 2007;64:1176–82. - PubMed
-
- Ueyama H, et al. Clinical heterogeneity in dysferlinopathy. Intern Med. 2002;41:532–6. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
