

Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies
- PMID: 21565291
- PMCID: PMC3146732
- DOI: 10.1016/j.ajhg.2011.04.013
Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies
Abstract
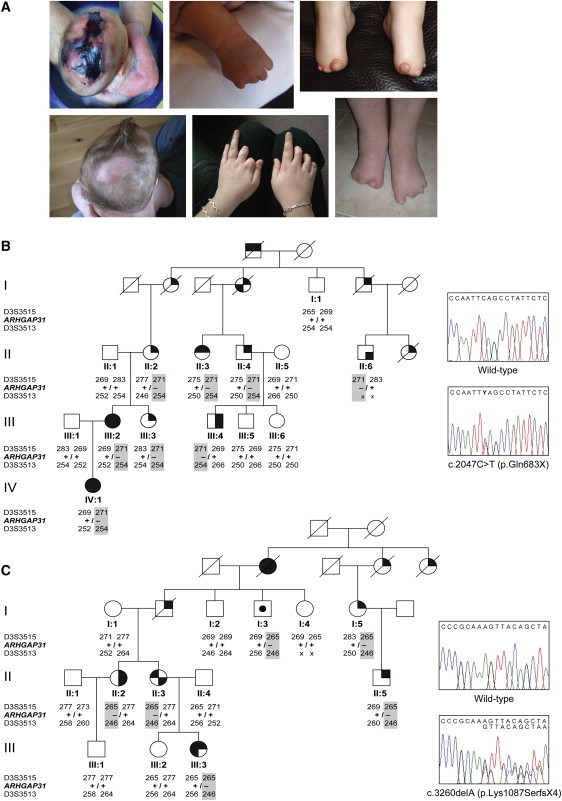
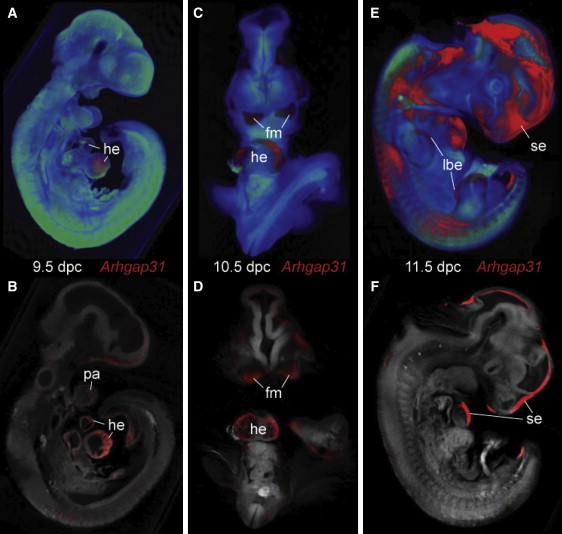
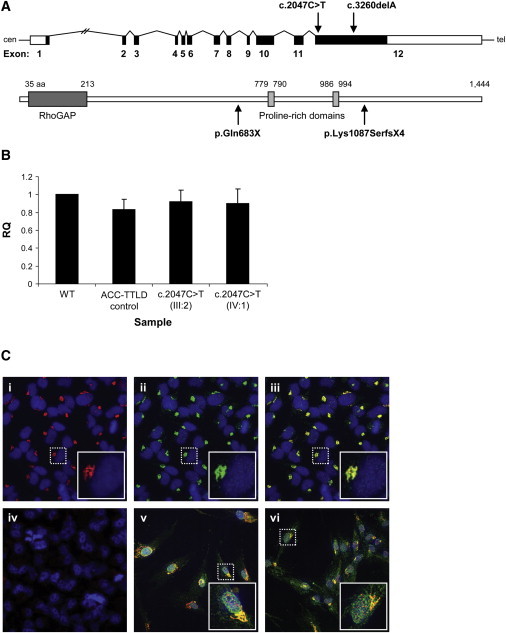
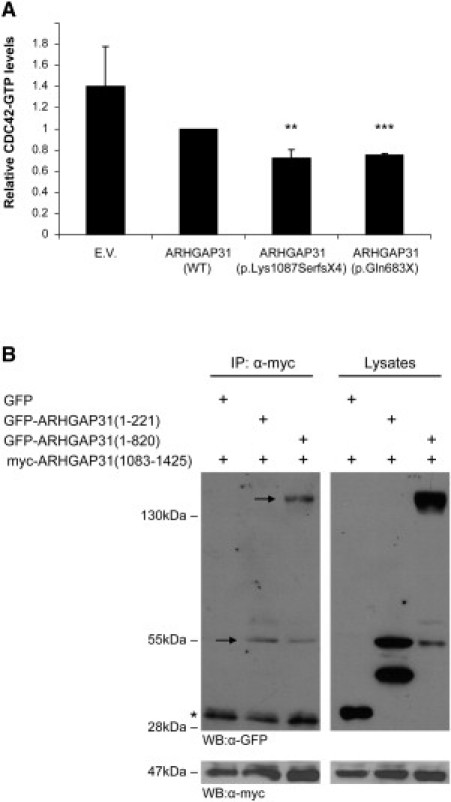
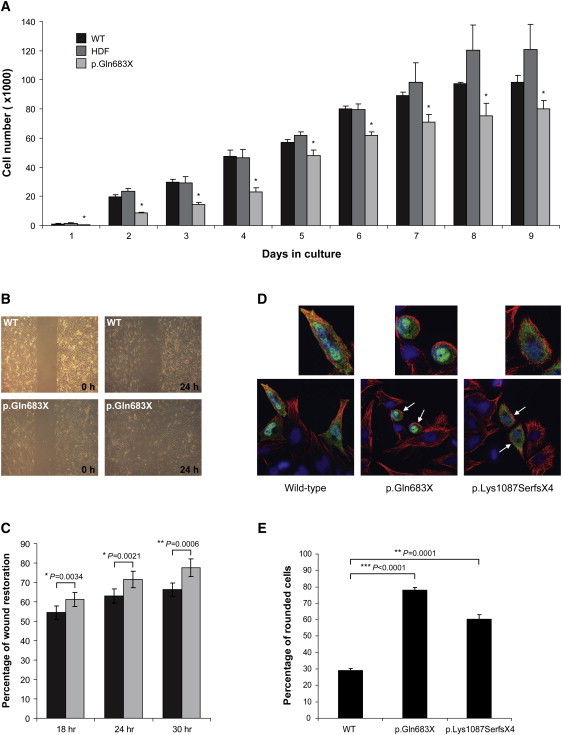
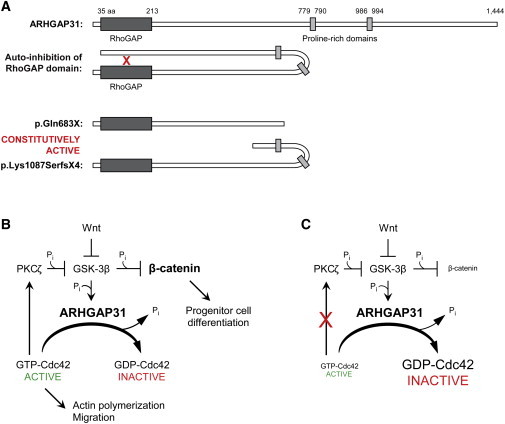
Regulation of cell proliferation and motility is essential for normal development. The Rho family of GTPases plays a critical role in the control of cell polarity and migration by effecting the cytoskeleton, membrane trafficking, and cell adhesion. We investigated a recognized developmental disorder, Adams-Oliver syndrome (AOS), characterized by the combination of aplasia cutis congenita (ACC) and terminal transverse limb defects (TTLD). Through a genome-wide linkage analysis, we detected a locus for autosomal-dominant ACC-TTLD on 3q generating a maximum LOD score of 4.93 at marker rs1464311. Candidate-gene- and exome-based sequencing led to the identification of independent premature truncating mutations in the terminal exon of the Rho GTPase-activating protein 31 gene, ARHGAP31, which encodes a Cdc42/Rac1 regulatory protein. Mutant transcripts are stable and increase ARHGAP31 activity in vitro through a gain-of-function mechanism. Constitutively active ARHGAP31 mutations result in a loss of available active Cdc42 and consequently disrupt actin cytoskeletal structures. Arhgap31 expression in the mouse is substantially restricted to the terminal limb buds and craniofacial processes during early development; these locations closely mirror the sites of impaired organogenesis that characterize this syndrome. These data identify the requirement for regulated Cdc42 and/or Rac1 signaling processes during early human development.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Tcherkezian J., Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell. 2007;99:67–86. - PubMed
-
- Vega F.M., Ridley A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101. - PubMed
-
- Snape K.M., Ruddy D., Zenker M., Wuyts W., Whiteford M., Johnson D., Lam W., Trembath R.C. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am. J. Med. Genet. A. 2009;149A:1860–1881. - PubMed
-
- Tcherkezian J., Triki I., Stenne R., Danek E.I., Lamarche-Vane N. The human orthologue of CdGAP is a phosphoprotein and a GTPase-activating protein for Cdc42 and Rac1 but not RhoA. Biol. Cell. 2006;98:445–456. - PubMed
-
- Verdyck P., Blaumeiser B., Holder-Espinasse M., Van Hul W., Wuyts W. Adams-Oliver syndrome: Clinical description of a four-generation family and exclusion of five candidate genes. Clin. Genet. 2006;69:86–92. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
