

Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies
- PMID: 21565292
- PMCID: PMC3146723
- DOI: 10.1016/j.ajhg.2011.04.014
Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies
Abstract
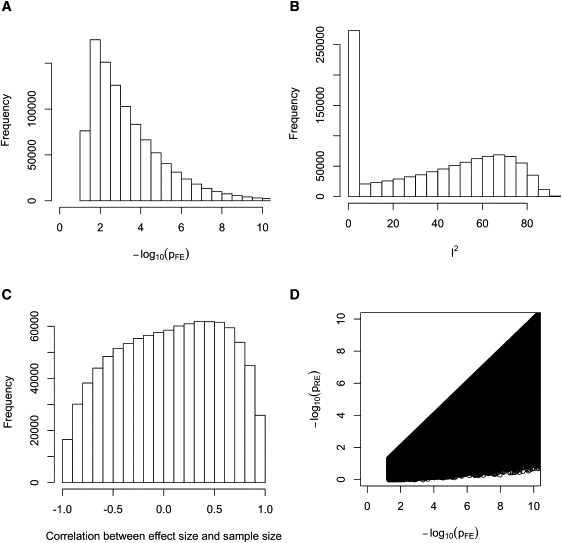
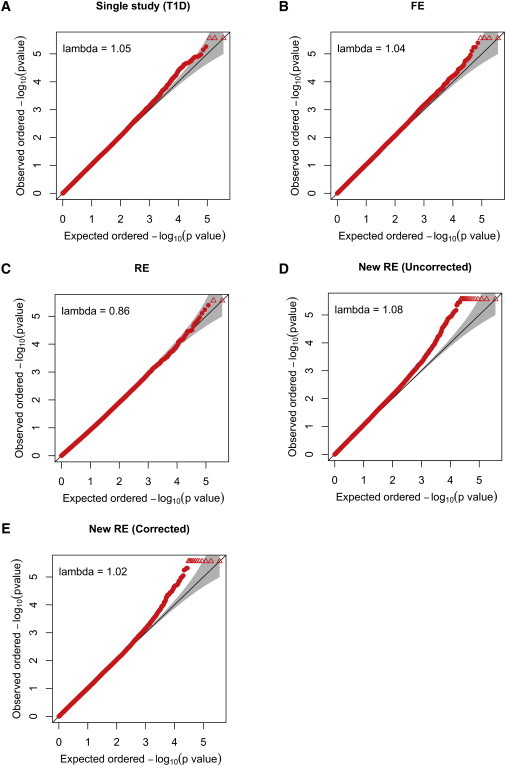
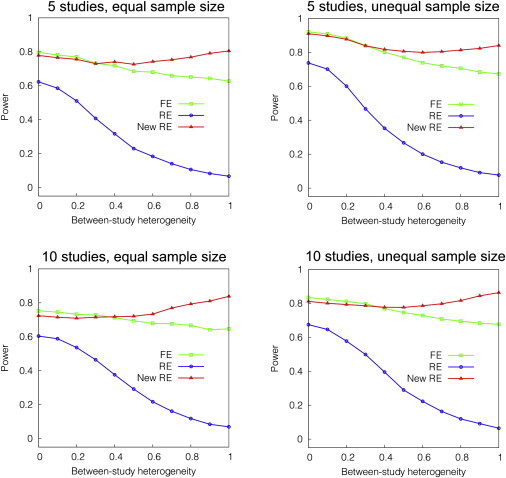
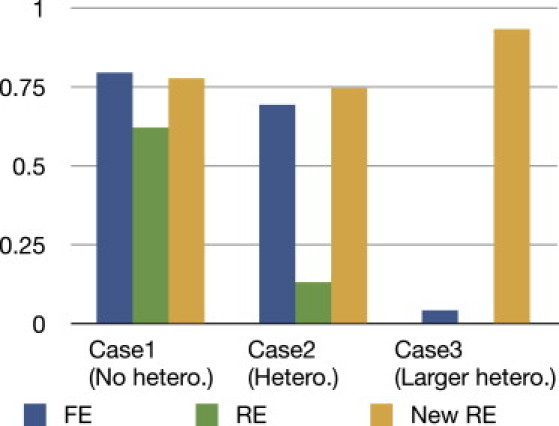
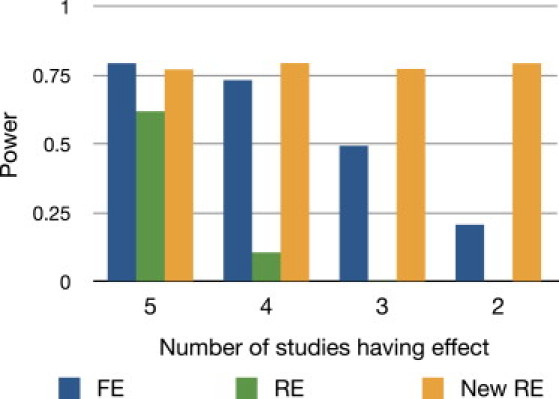
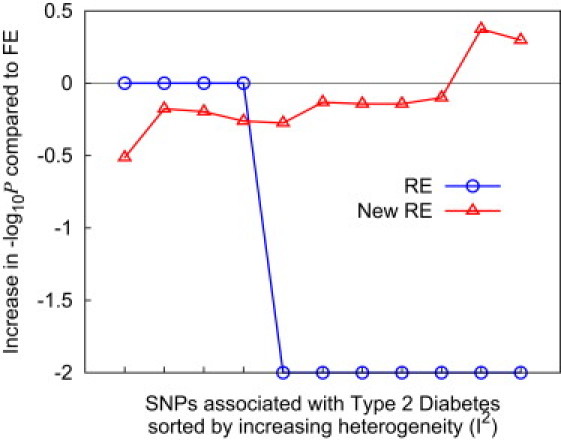
Meta-analysis is an increasingly popular tool for combining multiple different genome-wide association studies (GWASs) in a single aggregate analysis in order to identify associations with very small effect sizes. Because the data of a meta-analysis can be heterogeneous, referring to the differences in effect sizes between the collected studies, what is often done in the literature is to apply both the fixed-effects model (FE) under an assumption of the same effect size between studies and the random-effects model (RE) under an assumption of varying effect size between studies. However, surprisingly, RE gives less significant p values than FE at variants that actually show varying effect sizes between studies. This is ironic because RE is designed specifically for the case in which there is heterogeneity. As a result, usually, RE does not discover any associations that FE did not discover. In this paper, we show that the underlying reason for this phenomenon is that RE implicitly assumes a markedly conservative null-hypothesis model, and we present a new random-effects model that relaxes the conservative assumption. Unlike the traditional RE, the new method is shown to achieve higher statistical power than FE when there is heterogeneity, indicating that the new method has practical utility for discovering associations in the meta-analysis of GWASs.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P.A., Hirschhorn J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
