

Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression
- PMID: 21565614
- PMCID: PMC3259909
- DOI: 10.1016/j.cell.2011.03.035
Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression
Abstract
The molecular basis for p53-mediated tumor suppression remains unclear. Here, to elucidate mechanisms of p53 tumor suppression, we use knockin mice expressing an allelic series of p53 transcriptional activation mutants. Microarray analysis reveals that one mutant, p53(25,26), is severely compromised for transactivation of most p53 target genes, and, moreover, p53(25,26) cannot induce G(1)-arrest or apoptosis in response to acute DNA damage. Surprisingly, p53(25,26) retains robust activity in senescence and tumor suppression, indicating that efficient transactivation of the majority of known p53 targets is dispensable for these pathways. In contrast, the transactivation-dead p53(25,26,53,54) mutant cannot induce senescence or inhibit tumorigenesis, like p53 nullizygosity. Thus, p53 transactivation is essential for tumor suppression but, intriguingly, in association with a small set of novel p53 target genes. Together, our studies distinguish the p53 transcriptional programs involved in acute DNA-damage responses and tumor suppression-a critical goal for designing therapeutics that block p53-dependent side effects of chemotherapy without compromising p53 tumor suppression.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
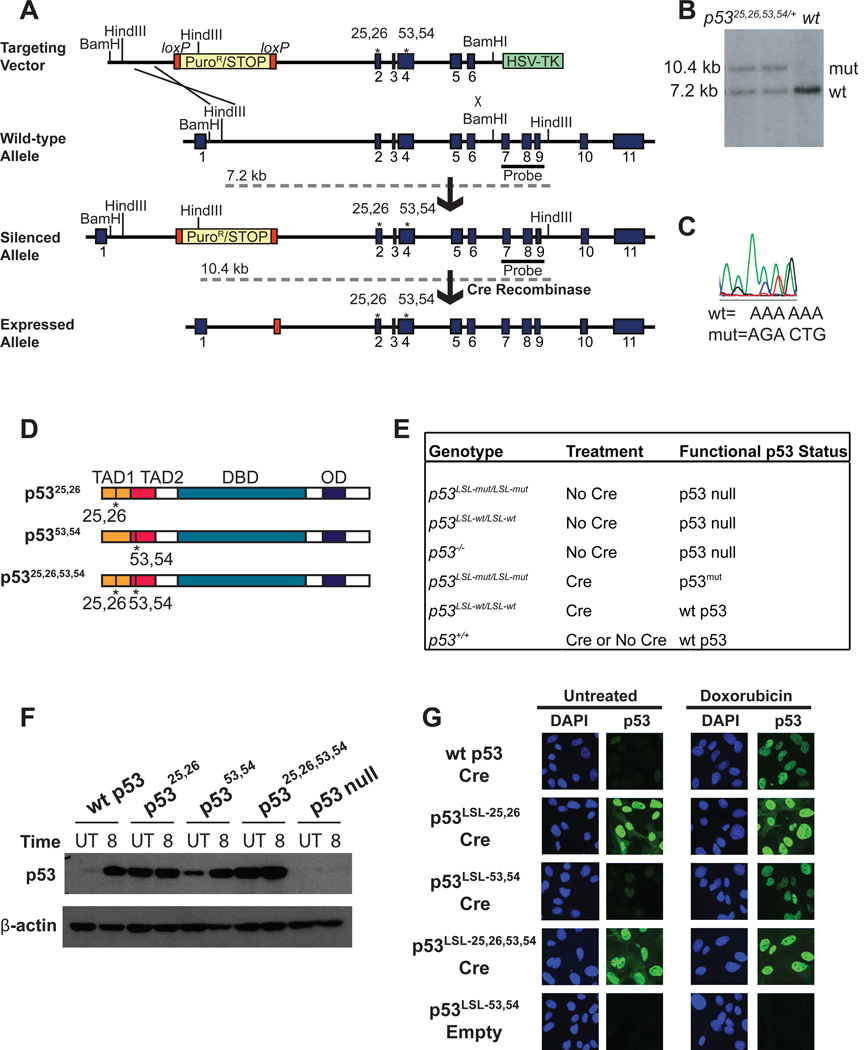
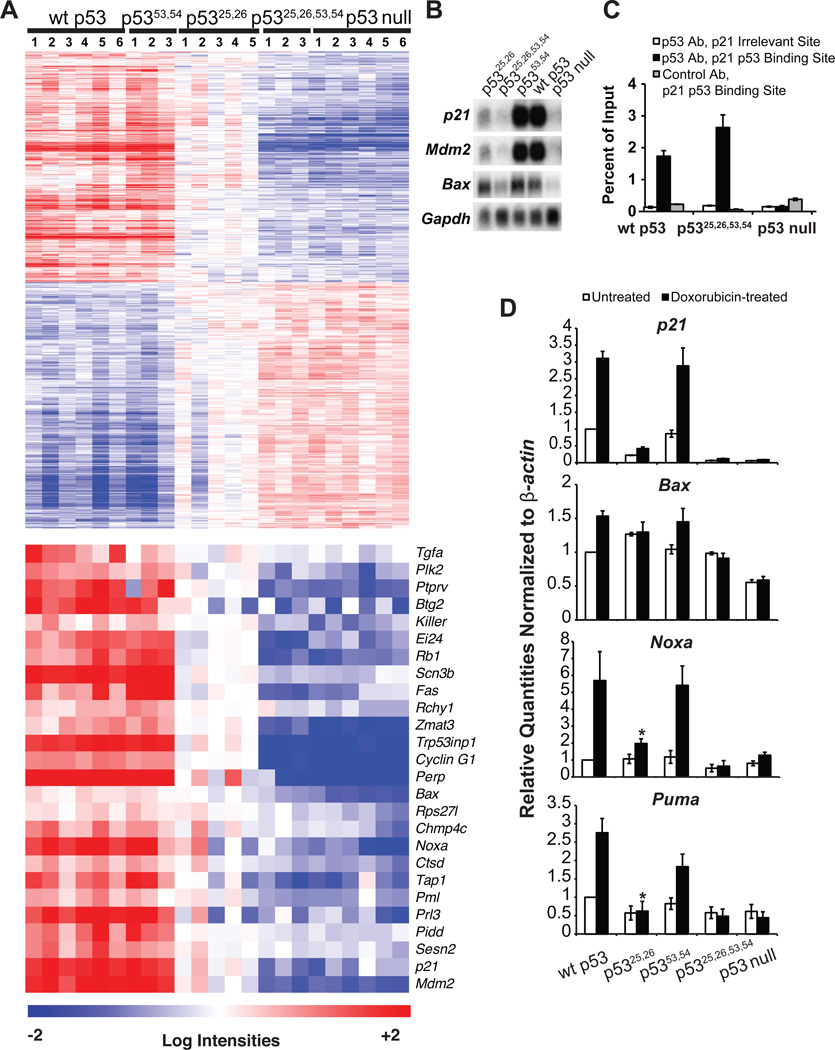
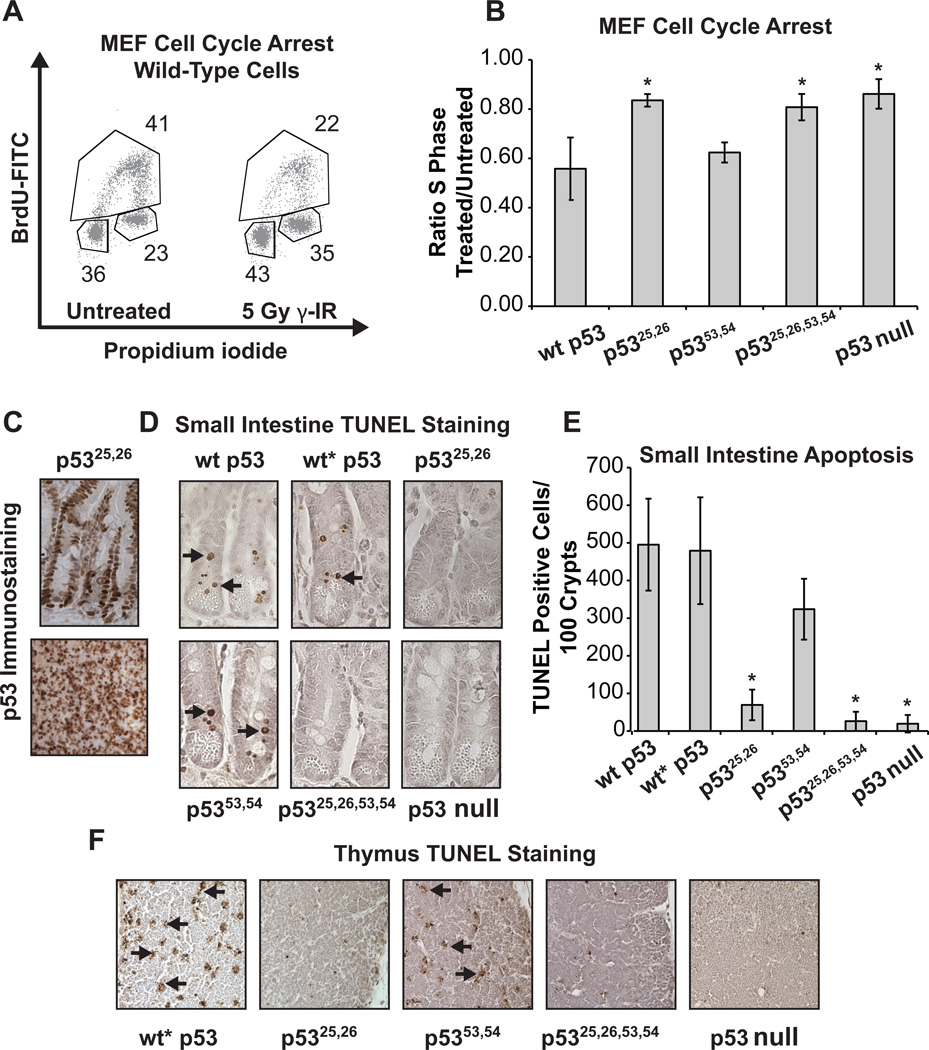
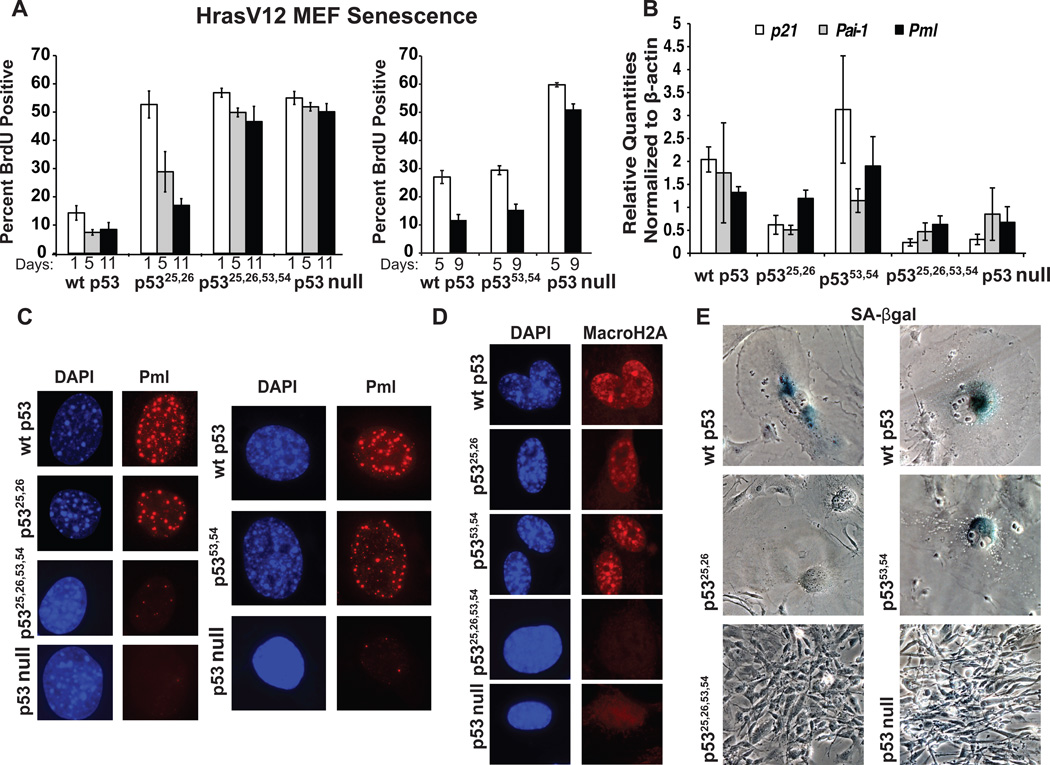
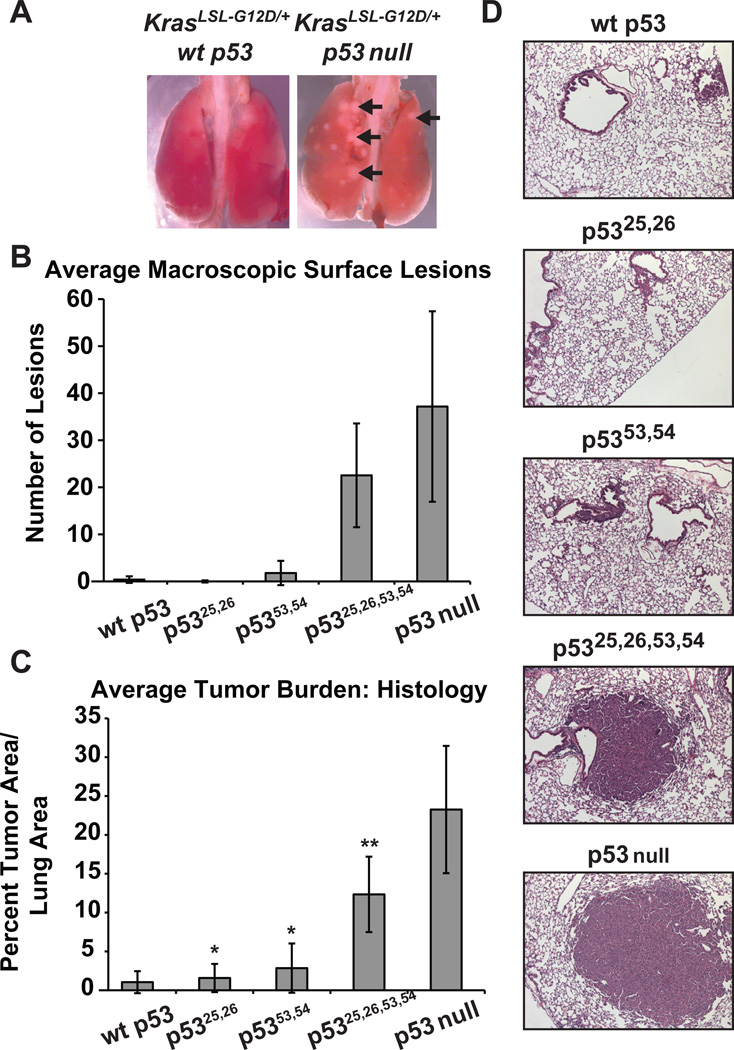
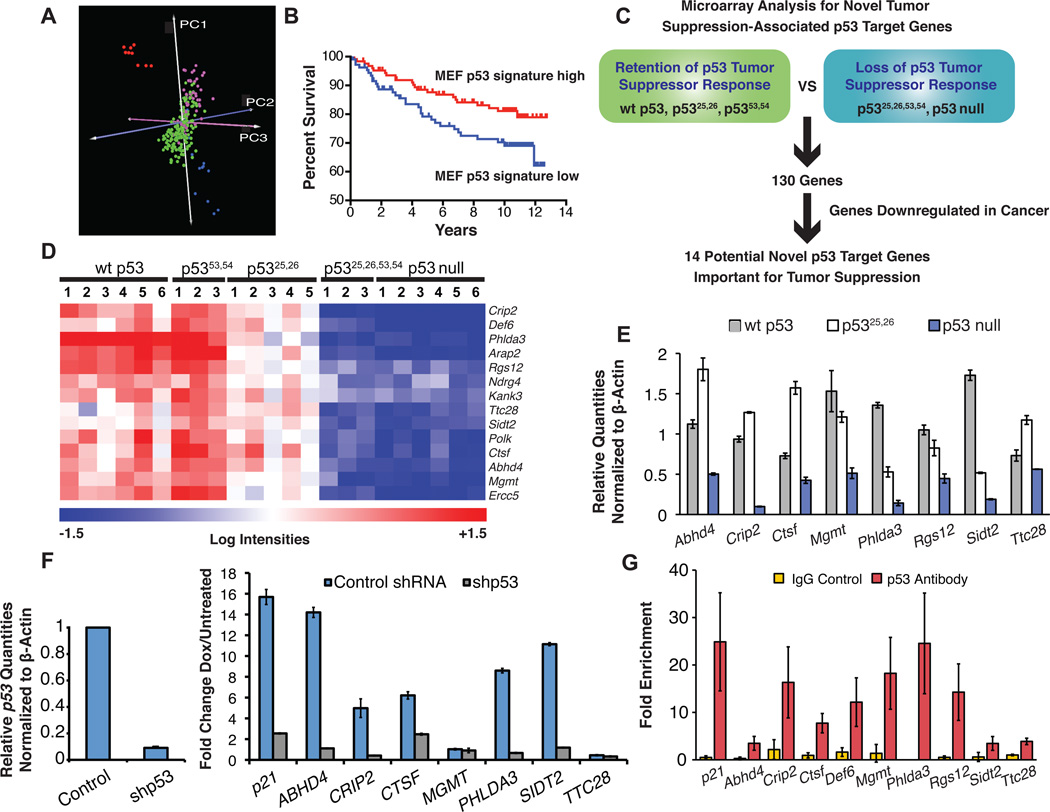
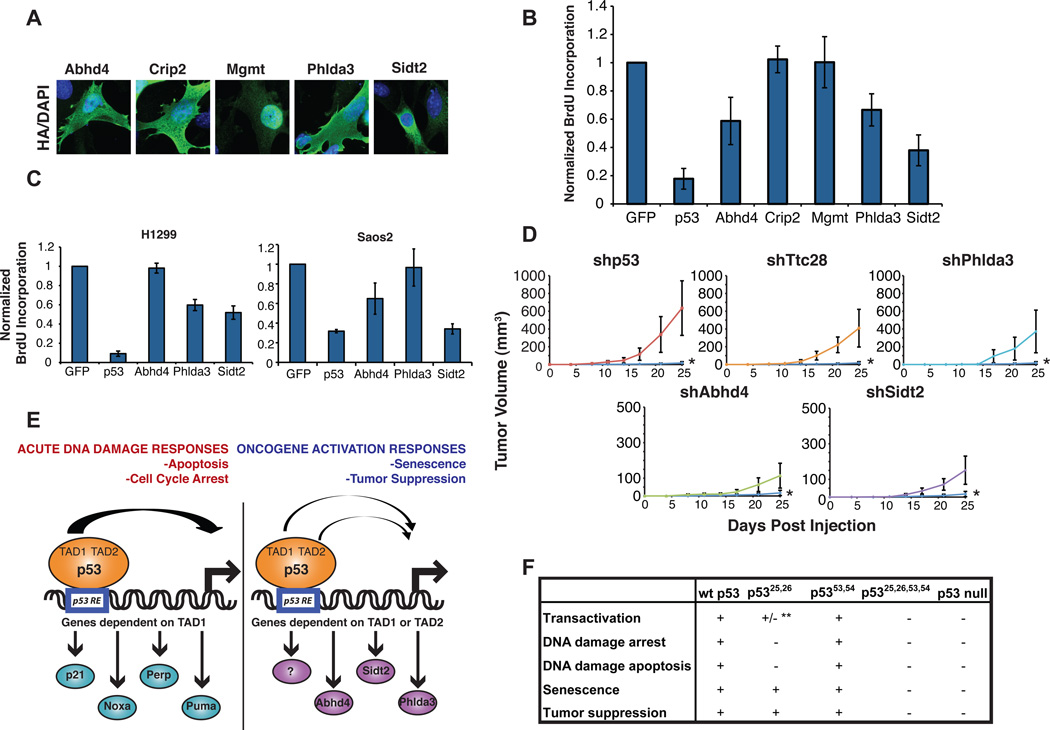
Comment in
-
Parsing p53 Transactivation.Dev Cell. 2011 May 17;20(5):573-4. doi: 10.1016/j.devcel.2011.04.015. Dev Cell. 2011. PMID: 21571212
References
-
- Attardi LD, de Vries A, Jacks T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene. 2004;23:973–980. - PubMed
-
- Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science (New York, NY. 1997;277:831–834. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
