

Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis
- PMID: 21569298
- PMCID: PMC3125325
- DOI: 10.1186/1750-1172-6-21
Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis
Abstract
Background: Usher syndrome (USH) combines sensorineural deafness with blindness. It is inherited in an autosomal recessive mode. Early diagnosis is critical for adapted educational and patient management choices, and for genetic counseling. To date, nine causative genes have been identified for the three clinical subtypes (USH1, USH2 and USH3). Current diagnostic strategies make use of a genotyping microarray that is based on the previously reported mutations. The purpose of this study was to design a more accurate molecular diagnosis tool.
Methods: We sequenced the 366 coding exons and flanking regions of the nine known USH genes, in 54 USH patients (27 USH1, 21 USH2 and 6 USH3).
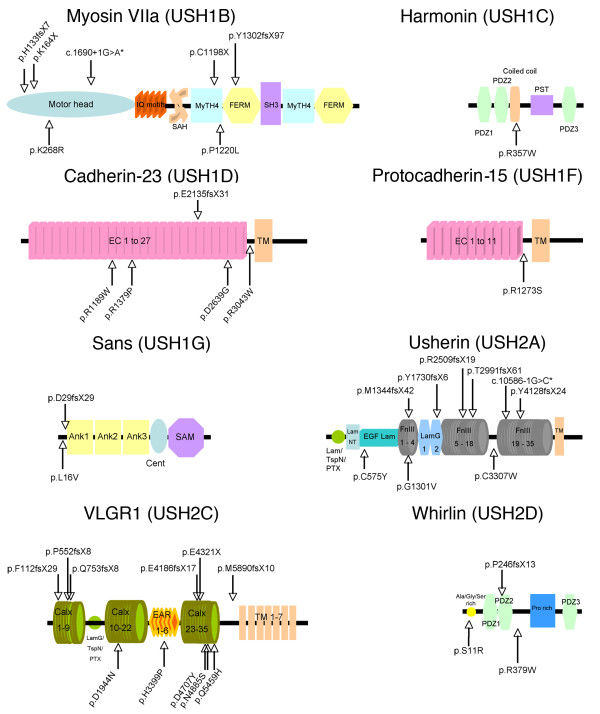
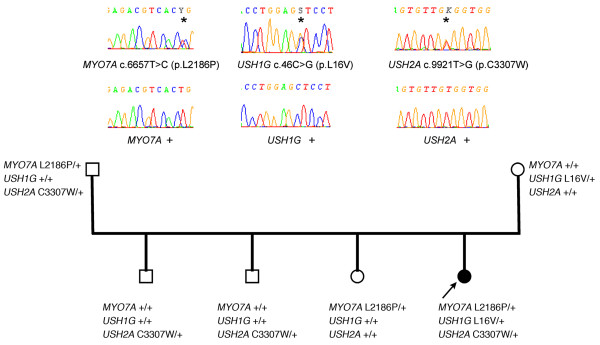
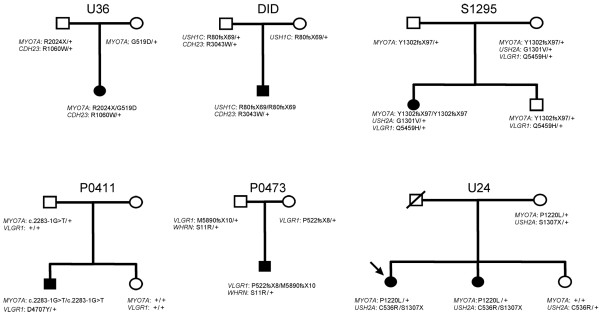
Results: Biallelic mutations were detected in 39 patients (72%) and monoallelic mutations in an additional 10 patients (18.5%). In addition to biallelic mutations in one of the USH genes, presumably pathogenic mutations in another USH gene were detected in seven patients (13%), and another patient carried monoallelic mutations in three different USH genes. Notably, none of the USH3 patients carried detectable mutations in the only known USH3 gene, whereas they all carried mutations in USH2 genes. Most importantly, the currently used microarray would have detected only 30 of the 81 different mutations that we found, of which 39 (48%) were novel.
Conclusions: Based on these results, complete exon sequencing of the currently known USH genes stands as a definite improvement for molecular diagnosis of this disease, which is of utmost importance in the perspective of gene therapy.
Figures

References
-
- Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/omim
-
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM. et al.A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet. 2000;26(1):56–60. doi: 10.1038/79178. - DOI - PubMed
-
- Verpy E, Leibovici M, Zwaenepoel I, Liu X-Z, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJB. et al.A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000;26(1):51–55. doi: 10.1038/79171. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
